Ryoichi Fukuda
Publications
Original article
| 57. |
Diels-Alder Cycloaddition of Cyclopentadiene and C60 at the Extreme High Pressure
T. Yang, R. Fukuda, R. Cammi, M. Ehara J. Phys. Chem. A 121, 4363-4371 (2017) + [Abstract] 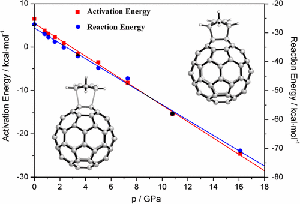 High-pressure Diels?Alder cycloaddition reaction of fullerenes is an important synthetic method for the thermally stable cycloadducts. The effects of high pressure on the potential energy surfaces of Diels-Alder cycloaddition of cyclopentadiene and C60 were studied with a recently developed approach, the polarizable continuum model for extreme pressure (XP-PCM). It is revealed that the high pressure reduces the activation energies and increases reaction energies drastically, making the DA reaction more favorable. The pressure effects on the reaction energetics can be divided into the cavitation and electronic contributions. For the activation energy, the cavitation contribution is significant in comparison with the electronic contribution. To assist future experiments, the activation volume and reaction volume were computed on the basis of the relationship between activation energy or reaction energy with the pressure as a consequence of the fitting linear correlation between activation energy or reaction energy with the pressure.
High-pressure Diels?Alder cycloaddition reaction of fullerenes is an important synthetic method for the thermally stable cycloadducts. The effects of high pressure on the potential energy surfaces of Diels-Alder cycloaddition of cyclopentadiene and C60 were studied with a recently developed approach, the polarizable continuum model for extreme pressure (XP-PCM). It is revealed that the high pressure reduces the activation energies and increases reaction energies drastically, making the DA reaction more favorable. The pressure effects on the reaction energetics can be divided into the cavitation and electronic contributions. For the activation energy, the cavitation contribution is significant in comparison with the electronic contribution. To assist future experiments, the activation volume and reaction volume were computed on the basis of the relationship between activation energy or reaction energy with the pressure as a consequence of the fitting linear correlation between activation energy or reaction energy with the pressure.
|
| 56. |
Core-Shell vs. Other Structures in Binary Cu38-nMn Nanocluster (M = Ru, Rh, Pd, Ag, Os, Ir, Pt, and Au; n = 1, 2, and 6): Theoretical Insight into Determining Factors
N. Takagi, K. Ishimura, M. Matsui, R. Fukuda, M. Ehara, S. Sakaki J. Phys. Chem. C 121, 10514-10528 (2017) + [Abstract] 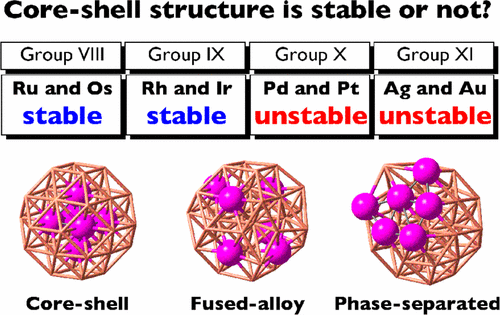 DFT calculations of binary transition-metal nanoclusters Cu38-nMn (M = Ru, Rh, Pd, Ag, Os, Ir, Pt, and Au; n = 1, 2, and 6) clearly show that a core-shell structure Cu32M6(core) with M in the core is stable for M = Ru, Rh, Os, and Ir but unstable for M = Pd, Ag, Pt, and Au. These results are consistent with the segregation energies evaluated for Cu37M. Electron population is more accumulated on the core M atoms in Cu38-nMn(core) (M = Ru, Rh, Os, and Ir) than on the core Cu atoms in Cu38. Such electron accumulation substantially occurs for M = Ru, Rh, Os, and Ir because the d orbitals of these transition metals are not fully occupied. A linear relationship was first found between the segregation energy and the increase in the d-orbital population of the core atom, indicating that the electron accumulation at the Mn core is one of the important factors for the segregation energy and the stabilization of the core-shell structure; in other words, a core-shell structure with M atom(s) in the core is stable when the d orbitals of M are not fully occupied. For M = Pd, Pt, and Au, the fused-alloy structure is more stable than the core-shell and phase-separated structures. For M = Ag, the fused-alloy structure is as stable as the phase-separated one but the core-shell structure is less stable. In these metals, the d orbitals are either nearly or fully occupied, and as a result, electron accumulation at the Mn core does not occur as much. For Cu32M6(core), the deformation energy of the Cu32 shell increases in the order Ru < Rh << Pd < Ag and Os < Ir << Pt < Au, because the size of the M6 core is substantially large for M = Pd, Ag, Pt, and Au. These results suggest that a large atom tends not to take the core position. The cohesive energies of Ru, Rh, Os, and Ir are larger than those of Pd, Ag, Pt, and Au, indicating that the cohesive energy is also an important property for understanding and discussing the structures of binary metal clusters/particles.
DFT calculations of binary transition-metal nanoclusters Cu38-nMn (M = Ru, Rh, Pd, Ag, Os, Ir, Pt, and Au; n = 1, 2, and 6) clearly show that a core-shell structure Cu32M6(core) with M in the core is stable for M = Ru, Rh, Os, and Ir but unstable for M = Pd, Ag, Pt, and Au. These results are consistent with the segregation energies evaluated for Cu37M. Electron population is more accumulated on the core M atoms in Cu38-nMn(core) (M = Ru, Rh, Os, and Ir) than on the core Cu atoms in Cu38. Such electron accumulation substantially occurs for M = Ru, Rh, Os, and Ir because the d orbitals of these transition metals are not fully occupied. A linear relationship was first found between the segregation energy and the increase in the d-orbital population of the core atom, indicating that the electron accumulation at the Mn core is one of the important factors for the segregation energy and the stabilization of the core-shell structure; in other words, a core-shell structure with M atom(s) in the core is stable when the d orbitals of M are not fully occupied. For M = Pd, Pt, and Au, the fused-alloy structure is more stable than the core-shell and phase-separated structures. For M = Ag, the fused-alloy structure is as stable as the phase-separated one but the core-shell structure is less stable. In these metals, the d orbitals are either nearly or fully occupied, and as a result, electron accumulation at the Mn core does not occur as much. For Cu32M6(core), the deformation energy of the Cu32 shell increases in the order Ru < Rh << Pd < Ag and Os < Ir << Pt < Au, because the size of the M6 core is substantially large for M = Pd, Ag, Pt, and Au. These results suggest that a large atom tends not to take the core position. The cohesive energies of Ru, Rh, Os, and Ir are larger than those of Pd, Ag, Pt, and Au, indicating that the cohesive energy is also an important property for understanding and discussing the structures of binary metal clusters/particles.
|
| 55. |
Comparing the Performance of TD-DFT and SAC-CI Methods in the Description of Excited States Potential Energy Surface: an Excited State Proton Transfer Reaction as Case Study
M. Savarese, U. Raucci, R. Fukuda, C. Adamo, M. Ehara, N. Rega, I. Ciofini J. Comput. Chem. 38, 1084-1092 (2017) + [Abstract] 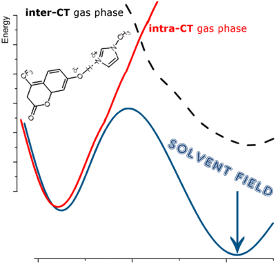 The performances, in the description of excited state potential energy surfaces, of several density functional approximations representative of the currently most applied exchange correlation functionals' families have been tested with respect to post Hartree-Fock references (here Symmetry Adapted Cluster-Configuration Interaction results). An experimentally well-characterized intermolecular proton transfer reaction has been considered as test case. The computed potential energy profiles were analyzed both in the gas phase and in toluene solution, here represented as a polarizable continuum model. The presence of intermolecular (dark) and intramolecular (bright) charge transfer excited states, whose polarity strongly differs along the reaction pathway, makes clear that only subtle compensation between spurious electronic effects -related to the incorrect asymptotic behavior of the functional- and solvent stabilization of polar states leads to the overall correct description of this excited state reaction when using global hybrids with low percentage of Hartree-Fock exchange.
The performances, in the description of excited state potential energy surfaces, of several density functional approximations representative of the currently most applied exchange correlation functionals' families have been tested with respect to post Hartree-Fock references (here Symmetry Adapted Cluster-Configuration Interaction results). An experimentally well-characterized intermolecular proton transfer reaction has been considered as test case. The computed potential energy profiles were analyzed both in the gas phase and in toluene solution, here represented as a polarizable continuum model. The presence of intermolecular (dark) and intramolecular (bright) charge transfer excited states, whose polarity strongly differs along the reaction pathway, makes clear that only subtle compensation between spurious electronic effects -related to the incorrect asymptotic behavior of the functional- and solvent stabilization of polar states leads to the overall correct description of this excited state reaction when using global hybrids with low percentage of Hartree-Fock exchange.
|
| 54. |
A Theoretical Investigation on CO Oxidation by Single-Atom Catalysts M1/γ-Al2O3 (M = Pd, Fe, Co, and Ni)
T. Yang, R. Fukuda, S. Hosokawa, T. Tanaka, S. Sakaki, M. Ehara ChemCatChem 9, 1222-1229 (2017) + [Abstract] 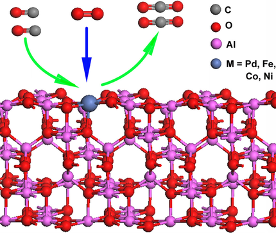 Single-atom catalysts have attracted much interest recently because of their excellent stability, high catalytic activity, and remarkable atom efficiency. Inspired by the recent experimental discovery of a highly efficient single-atom catalyst Pd1/γ-Al2O3, we conducted a comprehensive DFT study on geometries, stabilities and CO oxidation catalytic activities of M1/γ-Al2O3 (M=Pd, Fe, Co, and Ni) by using slab-model. One of the most important results here is that Ni1/Al2O3 catalyst exhibits higher activity in CO oxidation than Pd1/Al2O3. The CO oxidation occurs through the Mars van Krevelen mechanism, the rate-determining step of which is the generation of CO2 from CO through abstraction of surface oxygen. The projected density of states (PDOS) of 2p orbitals of the surface O, the structure of CO-adsorbed surface, charge polarization of CO and charge transfer from CO to surface are important factors for these catalysts. Although the binding energies of Fe and Co with Al2O3 are very large, those of Pd and Ni are small, indicating that the neighboring O atom is not strongly bound to Pd and Ni, which leads to an enhancement of the reactivity of the O atom toward CO. The metal oxidation state is suggested to be one of the crucial factors for the observed catalytic activity.
Single-atom catalysts have attracted much interest recently because of their excellent stability, high catalytic activity, and remarkable atom efficiency. Inspired by the recent experimental discovery of a highly efficient single-atom catalyst Pd1/γ-Al2O3, we conducted a comprehensive DFT study on geometries, stabilities and CO oxidation catalytic activities of M1/γ-Al2O3 (M=Pd, Fe, Co, and Ni) by using slab-model. One of the most important results here is that Ni1/Al2O3 catalyst exhibits higher activity in CO oxidation than Pd1/Al2O3. The CO oxidation occurs through the Mars van Krevelen mechanism, the rate-determining step of which is the generation of CO2 from CO through abstraction of surface oxygen. The projected density of states (PDOS) of 2p orbitals of the surface O, the structure of CO-adsorbed surface, charge polarization of CO and charge transfer from CO to surface are important factors for these catalysts. Although the binding energies of Fe and Co with Al2O3 are very large, those of Pd and Ni are small, indicating that the neighboring O atom is not strongly bound to Pd and Ni, which leads to an enhancement of the reactivity of the O atom toward CO. The metal oxidation state is suggested to be one of the crucial factors for the observed catalytic activity.
|
| 53. |
Structures of bimetallic copper-ruthenium nanoparticles: incoherent interface and surface active sites for catalytic nitric oxide dissociation
R. Fukuda, N. Takagi, S. Sakaki, M. Ehara J. Phys. Chem. C 121, 300-307 (2017) + [Abstract] 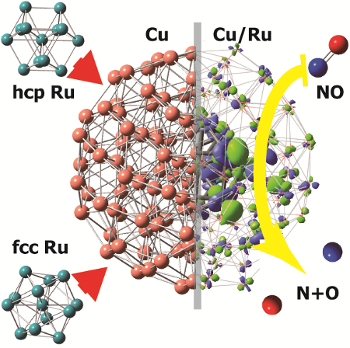 Bimetallic alloy nanoparticles are promising candidates for replacing platinum group metals utilized in the catalytic removal of nitrogen oxides in exhaust gas. In this study, we investigated the electronic, interfacial, and surface structures of copper/ruthenium alloy nanoparticles by quantum chemical computations using 135-atomic cluster models. We employed Ru-core/Cu-shell models in which the Ru-core takes both fcc (face-centered cubic) and hcp (hexagonal closed-packed) structures. The fcc-core model has a coherent Cu/Ru interface, while the hcp-core model involves an incoherent interface. This incoherence results in discontinuity in the lattice structure and the valence electronic structure, and generates step-like structures on the surface of the particle. Such a step-like site enhances the catalytic activities for nitric oxide dissociation. The orbital energies suggest that the alloying can control the oxidation tendency of clusters. Charge-transfer occurs between the Cu shell and Ru core; the surface layer of the clusters has a positive charge, although the surface atoms are not directly binding to the core Ru atoms. The interfacial structure of core?shell interphase is a crucial factor to be considered in designing the properties of alloy nanoparticles.
Bimetallic alloy nanoparticles are promising candidates for replacing platinum group metals utilized in the catalytic removal of nitrogen oxides in exhaust gas. In this study, we investigated the electronic, interfacial, and surface structures of copper/ruthenium alloy nanoparticles by quantum chemical computations using 135-atomic cluster models. We employed Ru-core/Cu-shell models in which the Ru-core takes both fcc (face-centered cubic) and hcp (hexagonal closed-packed) structures. The fcc-core model has a coherent Cu/Ru interface, while the hcp-core model involves an incoherent interface. This incoherence results in discontinuity in the lattice structure and the valence electronic structure, and generates step-like structures on the surface of the particle. Such a step-like site enhances the catalytic activities for nitric oxide dissociation. The orbital energies suggest that the alloying can control the oxidation tendency of clusters. Charge-transfer occurs between the Cu shell and Ru core; the surface layer of the clusters has a positive charge, although the surface atoms are not directly binding to the core Ru atoms. The interfacial structure of core?shell interphase is a crucial factor to be considered in designing the properties of alloy nanoparticles.
|
| 52. |
Electronic Transitions in Confomationally Controlled Peralkylated Hexasilanes
Y. Kanazawa, H. Tsuji, M. Ehara, R. Fukuda, D. L. Casher, K. Tamao, H. Nakatsuji, J. Michl, ChemPhysChem 19, 3010-3022 (2016) + [Abstract]  The photophysical properties of oligosilanes show unique conformational dependence due to σ-electron delocalization. The excited states of the SAS, AAS, and AEA conformations of peralkylated n-hexasilanes, in which the SiSiSiSi dihedral angles are controlled into a syn (S), anti (A), or eclipsed (E) conformation, were investigated by using UV absorption, magnetic circular dichroism (MCD), and linear dichroism spectroscopy. Simultaneous Gaussian fitting of all three spectra identified a minimal set of transitions and the wavenumbers, oscillator strengths, and MCD B terms in all three compounds. The results compare well with those obtained by using the symmetry-adapted-cluster configuration interaction method and almost as well with those obtained by time-dependent density functional theory with the PBE0 functional. The conformational dependence of the transition energies and other properties of free-chain permethylated n-hexasilane, n-Si6Me14, was also examined as a function of dihedral angles, and the striking effects found were attributed to avoided crossings between configurations of σσ* and σπ* character.
The photophysical properties of oligosilanes show unique conformational dependence due to σ-electron delocalization. The excited states of the SAS, AAS, and AEA conformations of peralkylated n-hexasilanes, in which the SiSiSiSi dihedral angles are controlled into a syn (S), anti (A), or eclipsed (E) conformation, were investigated by using UV absorption, magnetic circular dichroism (MCD), and linear dichroism spectroscopy. Simultaneous Gaussian fitting of all three spectra identified a minimal set of transitions and the wavenumbers, oscillator strengths, and MCD B terms in all three compounds. The results compare well with those obtained by using the symmetry-adapted-cluster configuration interaction method and almost as well with those obtained by time-dependent density functional theory with the PBE0 functional. The conformational dependence of the transition energies and other properties of free-chain permethylated n-hexasilane, n-Si6Me14, was also examined as a function of dihedral angles, and the striking effects found were attributed to avoided crossings between configurations of σσ* and σπ* character.
|
| 51. |
Electronic excitation and ionization behavior of N-hydroxypyridine-2(1H)-thione and its deprotonated anion in a polarizable medium studied using quantum chemical computations
R. Fukuda, and M. Ehara Theor. Chem. Acc. 135, 105 (9 pages) (2016) + [Abstract] 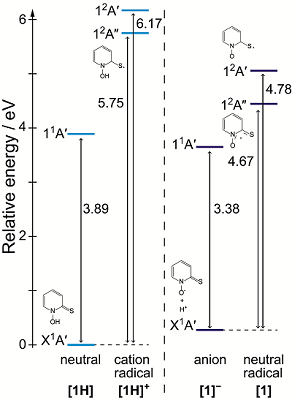 N-Hydroxypyridine-2(1H)-thione (N-HPT) is an important photochemical generator of hydroxyl radicals; however, it has been pointed out that N-HPT is not a specific precursor of hydroxyl radical. Photoionization of N-HPT competes with photochemical N-O bond cleavage in neutral aqueous solution. The possibility of a competitive reaction could be critical for studies using N-HPT as the radical precursor; therefore, the detailed behaviors of electronic excitation and ionization of N-HPT and its deprotonated anion, which is the dominant tautomer under neutral pH conditions, are studied using quantum chemical methods with the symmetry-adapted cluster-configuration interaction (SAC-CI) method and the polarizable continuum model (PCM). The detailed assignment of the UV-vis spectra of N-HPT is provided, and the origin of the observed negative solvatochromism is found to be the charge transfer excitation between the sulfur and the pyridine ring. The photochemical N-O bond cleavage occurs via the conical intersections between the lowest π→π* and π→σ* states and between the π→σ* and ground state, when N-HPT dissociates into PyS· and ·OH radicals. The calculated ionization potentials of N-HPT and the deprotonated N-HPT anion are 5.75 and 4.67 eV in PCM water. This demonstrates that the charge transfer excitation energy between N-HPT and liquid water becomes significantly lower for the deprotonated anion in comparison with the neutral molecule. Even under mild photochemical conditions, photoinduced ionization of N-HPT may occur in neutral aqueous solution.
N-Hydroxypyridine-2(1H)-thione (N-HPT) is an important photochemical generator of hydroxyl radicals; however, it has been pointed out that N-HPT is not a specific precursor of hydroxyl radical. Photoionization of N-HPT competes with photochemical N-O bond cleavage in neutral aqueous solution. The possibility of a competitive reaction could be critical for studies using N-HPT as the radical precursor; therefore, the detailed behaviors of electronic excitation and ionization of N-HPT and its deprotonated anion, which is the dominant tautomer under neutral pH conditions, are studied using quantum chemical methods with the symmetry-adapted cluster-configuration interaction (SAC-CI) method and the polarizable continuum model (PCM). The detailed assignment of the UV-vis spectra of N-HPT is provided, and the origin of the observed negative solvatochromism is found to be the charge transfer excitation between the sulfur and the pyridine ring. The photochemical N-O bond cleavage occurs via the conical intersections between the lowest π→π* and π→σ* states and between the π→σ* and ground state, when N-HPT dissociates into PyS· and ·OH radicals. The calculated ionization potentials of N-HPT and the deprotonated N-HPT anion are 5.75 and 4.67 eV in PCM water. This demonstrates that the charge transfer excitation energy between N-HPT and liquid water becomes significantly lower for the deprotonated anion in comparison with the neutral molecule. Even under mild photochemical conditions, photoinduced ionization of N-HPT may occur in neutral aqueous solution.
|
| 50. |
Projected CAP/SAC -CI method with smooth Voronoi potential for calculating resonance states
M. Ehara, R. Fukuda, and T. Sommerfeld J. Comput. Chem. 37, 242-249 (2016) + [Abstract] 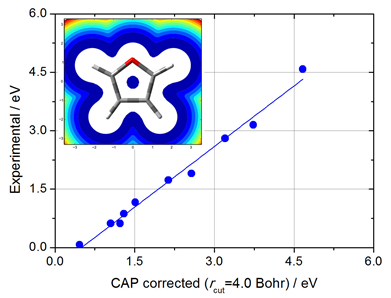 The complex absorbing potential (CAP)/symmetry-adapted cluster-configuration interaction (SAC-CI) method has been combined with a smooth Voronoi potential, which was recently introduced in the extrapolation procedure, to locate π* resonance states of small- to medium-size molecules. Here, the projected CAP/SAC-CI method is combined with this potential and used to calculate the double-bond and heteroaromatic π* resonances of acetaldehyde, butadiene, glyoxal, pyridine, pyrazine, and furan. As observed in the pilot applications, the corrected η-trajectories provide a stable resonance energy and width-or lifetime-regardless of the size parameter (rcut) of the smooth Voronoi potential. However, in general, the stabilization behavior of the trajectories is clearer for larger rcut values, which implies that the interaction of the CAP with the valence electrons is more advantageously addressed by a larger "cavity" size.
The complex absorbing potential (CAP)/symmetry-adapted cluster-configuration interaction (SAC-CI) method has been combined with a smooth Voronoi potential, which was recently introduced in the extrapolation procedure, to locate π* resonance states of small- to medium-size molecules. Here, the projected CAP/SAC-CI method is combined with this potential and used to calculate the double-bond and heteroaromatic π* resonances of acetaldehyde, butadiene, glyoxal, pyridine, pyrazine, and furan. As observed in the pilot applications, the corrected η-trajectories provide a stable resonance energy and width-or lifetime-regardless of the size parameter (rcut) of the smooth Voronoi potential. However, in general, the stabilization behavior of the trajectories is clearer for larger rcut values, which implies that the interaction of the CAP with the valence electrons is more advantageously addressed by a larger "cavity" size.
|
| 49. |
Exploring excited states using time dependent density functional theory and density-based indexes C. Adamo, T. Le Bahers, M. Savarese, L. Wilbraham, G. García, R. Fukuda, M. Ehara, N. Rega, and I. Ciofini Coord. Chem. Rev. 304-305 , 166-178 (2015) + [Abstract] 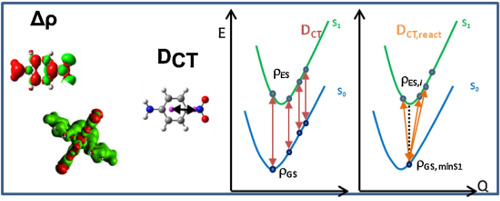 The recent advances in the development and application of density-based indexes for the description of the nature and the quantification of the extent of charge transfer associated with a given electronic transition are here reviewed. Starting from the basic definition of the indexes, a brief overview of their potential as indicators of potentially problematic cases in the description of charge transfer excitations using Time Dependent Density Functional Theory (TD-DFT) will be first given together with their possible application for comparing TD-DFT results to post Hartree-Fock (post-HF) calculations. After this methodological part, several examples of the application of density-based indexes to describe, from a quantitative and qualitative point of view, the charge transfer character (for instance in push-pull systems) or to map excited state reaction pathways (for instance in the case of Excited State Proton Transfer reactions) will be given to exemplify the insights that these indexes may bring to the description and design of new compounds of potential technological relevance.
The recent advances in the development and application of density-based indexes for the description of the nature and the quantification of the extent of charge transfer associated with a given electronic transition are here reviewed. Starting from the basic definition of the indexes, a brief overview of their potential as indicators of potentially problematic cases in the description of charge transfer excitations using Time Dependent Density Functional Theory (TD-DFT) will be first given together with their possible application for comparing TD-DFT results to post Hartree-Fock (post-HF) calculations. After this methodological part, several examples of the application of density-based indexes to describe, from a quantitative and qualitative point of view, the charge transfer character (for instance in push-pull systems) or to map excited state reaction pathways (for instance in the case of Excited State Proton Transfer reactions) will be given to exemplify the insights that these indexes may bring to the description and design of new compounds of potential technological relevance.
|
| 48. |
Synthesis and optical properties of imidazole and benzimidazole-based fused π-conjugated compounds: Influence of substituent, counter anion, and π-conjugated system
K. Takagi, K. Kusafuka, Y. Ito. K. Yamauchi, K. Ito, R. Fukuda, and M. Ehara J. Org. Chem. 80, 7172-7183 (2015) + [Abstract] 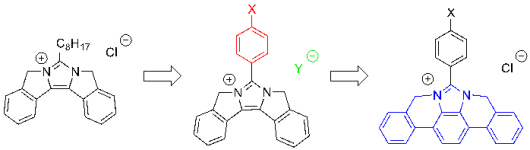 Fused π-conjugated imidazolium chlorides having the hydrogen (1-Cl), octyloxy (2-Cl), N,Ndibutylamino (3-Cl), trifluoromethyl (4-Cl), and cyano (5-Cl) groups-substituted benzene ring at the 2- position of imidazole were prepared. The counter anion exchanges from chloride to bis(trifluoromethanesulfonyl)imidate (2-TFSI) and tetrafluoroborate (2-BF4) were performed. The optical properties of these compounds (absorption and emission wavelengths, fluorescence quantum yield, and solvatochromism) were influenced by both the substituent and anion character, which was investigated by the theoretical calculations using the density functional theory (DFT) and symmetryadapted cluster-configuration interaction (SAC-CI) methods. Fused π-conjugated benzimidazolium chlorides having N,N-dibutylamino (6-Cl) and cyano (7-Cl) groups were also prepared to observe the different solvatochromic shifts.
Fused π-conjugated imidazolium chlorides having the hydrogen (1-Cl), octyloxy (2-Cl), N,Ndibutylamino (3-Cl), trifluoromethyl (4-Cl), and cyano (5-Cl) groups-substituted benzene ring at the 2- position of imidazole were prepared. The counter anion exchanges from chloride to bis(trifluoromethanesulfonyl)imidate (2-TFSI) and tetrafluoroborate (2-BF4) were performed. The optical properties of these compounds (absorption and emission wavelengths, fluorescence quantum yield, and solvatochromism) were influenced by both the substituent and anion character, which was investigated by the theoretical calculations using the density functional theory (DFT) and symmetryadapted cluster-configuration interaction (SAC-CI) methods. Fused π-conjugated benzimidazolium chlorides having N,N-dibutylamino (6-Cl) and cyano (7-Cl) groups were also prepared to observe the different solvatochromic shifts.
|
| 47. |
How can we understand Au8 cores and entangled ligands of selenolate- and stThiolate-protected gold nanoclusters Au24(ER)20 and Au20(ER)16 (E = Se, S; R = Ph, Me)? A theoretical study
N. Takagi, K. Ishimura, R. Fukuda, T. Matsui, T. Nakajima, M. Ehara, and S. Sakaki J. Am. Chem. Soc. 137, 8593-8602 (2015) + [Abstract] 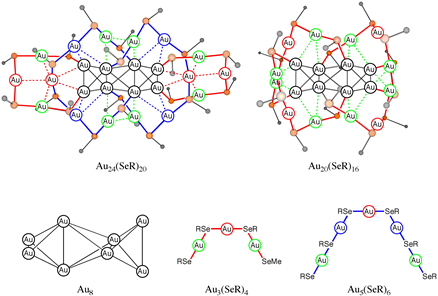 The geometries and electronic structures of selenolate-protected Au nanoclusters, Au24(SeR)20 and Au20(SeR)16, and their thiolate analogues are theoretically investigated with DFT and SCS-MP2 methods, to elucidate the electronic structure of their unusual Au8 core and the reason why they have the abnormal entangled geometry of "staple-like" chain ligands. The Au8 core is understood to be an [Au4]2+ dimer in which the [Au4]2+ species has a tetrahedral geometry with a closed-shell singlet electronic structure. The SCS-MP2 method successfully reproduced the distance between two [Au4]2+ species but the DFT with various functionals failed, suggesting that the dispersion interaction is crucial between these two [Au4]2+ species. The SCS-MP2-calculated formation energies of these nanocluster compounds indicate that the thiolate staple-like chain ligands are more stable than the selenolate ones but the Au8 core more strongly coordinates with the selenolate staple-like chain ligands than with the thiolate ones. Though Au20(SeR)16 has not been reported yet, its formation energy is calculated to be large, suggesting that this compound can be synthesized as a stable species. The aurophilic interaction between the staple-like chain ligands and between the Au8 core and the staple-like chain ligand plays an important role for the total stability. This is one of the important reasons why the unusual entangled ligands are involved in these compounds.
The geometries and electronic structures of selenolate-protected Au nanoclusters, Au24(SeR)20 and Au20(SeR)16, and their thiolate analogues are theoretically investigated with DFT and SCS-MP2 methods, to elucidate the electronic structure of their unusual Au8 core and the reason why they have the abnormal entangled geometry of "staple-like" chain ligands. The Au8 core is understood to be an [Au4]2+ dimer in which the [Au4]2+ species has a tetrahedral geometry with a closed-shell singlet electronic structure. The SCS-MP2 method successfully reproduced the distance between two [Au4]2+ species but the DFT with various functionals failed, suggesting that the dispersion interaction is crucial between these two [Au4]2+ species. The SCS-MP2-calculated formation energies of these nanocluster compounds indicate that the thiolate staple-like chain ligands are more stable than the selenolate ones but the Au8 core more strongly coordinates with the selenolate staple-like chain ligands than with the thiolate ones. Though Au20(SeR)16 has not been reported yet, its formation energy is calculated to be large, suggesting that this compound can be synthesized as a stable species. The aurophilic interaction between the staple-like chain ligands and between the Au8 core and the staple-like chain ligand plays an important role for the total stability. This is one of the important reasons why the unusual entangled ligands are involved in these compounds.
|
| 46. |
Modeling molecular systems at extreme pressure by an extension of the polarizable continuum model (PCM) based on the symmetry-adapted cluster-configuration interaction (SAC-CI) method: confined electronic excited states of furan as a test case
R. Fukuda, M. Ehara, and R. Cammi J. Chem. Theory Comput. 11, 2063-2076 (2015) + [Abstract] 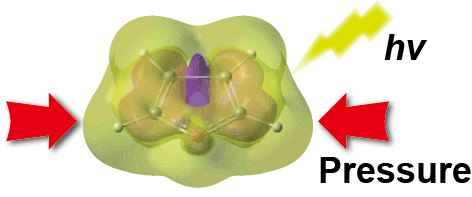 Novel molecular photochemistry can be developed by combining the high pressure and laser irradiation. For studying such high-pressure effects on the confined electronic ground and excited states, we extend the PCM (polarizable continuum model) SAC (symmetry-adapted cluster) and SAC-CI (SAC-configuration interaction) methods to the PCM-XP (extreme pressure) framework. With using the PCM-XP SAC/SAC-CI method, molecular systems in various electronic states can be confined by the polarizable media in a smooth and flexible way. The PCM-XP SAC/SAC-CI method is applied to furan (C4H4O) molecule in cyclohexane at high pressure (1-60 GPa). The relation between the calculated free-energy and cavity volume can be approximately represented with the Murnaghan equation of state. The excitation energies of furan in cyclohexane show blueshifts with increasing the pressure, and the extents of the blueshifts significantly depend on the character of the excitations. Particularly large confinement effects are found in the Rydberg states. The energy ordering of the lowest Rydberg and valence states alters under high-pressure. The pressure effects on the electronic structure may be classified into two contributions: a confinement of molecular orbital and a suppression of the mixing between the valence and Rydberg configurations. The valence or Rydberg character in an excited state is, therefore, enhanced under high pressure.
Novel molecular photochemistry can be developed by combining the high pressure and laser irradiation. For studying such high-pressure effects on the confined electronic ground and excited states, we extend the PCM (polarizable continuum model) SAC (symmetry-adapted cluster) and SAC-CI (SAC-configuration interaction) methods to the PCM-XP (extreme pressure) framework. With using the PCM-XP SAC/SAC-CI method, molecular systems in various electronic states can be confined by the polarizable media in a smooth and flexible way. The PCM-XP SAC/SAC-CI method is applied to furan (C4H4O) molecule in cyclohexane at high pressure (1-60 GPa). The relation between the calculated free-energy and cavity volume can be approximately represented with the Murnaghan equation of state. The excitation energies of furan in cyclohexane show blueshifts with increasing the pressure, and the extents of the blueshifts significantly depend on the character of the excitations. Particularly large confinement effects are found in the Rydberg states. The energy ordering of the lowest Rydberg and valence states alters under high-pressure. The pressure effects on the electronic structure may be classified into two contributions: a confinement of molecular orbital and a suppression of the mixing between the valence and Rydberg configurations. The valence or Rydberg character in an excited state is, therefore, enhanced under high pressure.
|
| 45. |
Proton-induced generation of remote N-heterocyclic carbene-Ru complexes
T. Fukushima, R. Fukuda, K. Kobayashi, G. F. Caramori, G. Frenking, M. Ehara,and K. Tanaka Chem. Euro. J. 21, 106-110 (2015) + [Abstract] 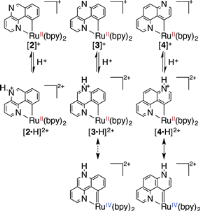 The proton induced Ru-C bond variation which was previously found to be relevant in the water oxidation has been investigated using cyclometalated ruthenium complexes with three phenanthroline (phen) isomers. The designed complexes, [Ru(bpy)2(1,5-phen)]+ ([2]+), [Ru(bpy)2(1,6-phen)]+ ([3]+), and [Ru(bpy)2(1,7-phen)]+ ([4]+) were newly synthesized and their structural and electronic properties were analyzed by various spectroscopy and theoretical protocol. Protonation of [4]+ triggered profound electronic structural change to form remote N-heterocyclic carbene (rNHC), while protonation of [2]+ and [3]+ did not affect their structures. It was found that changes in the electronic structure of phen beyond classical resonance forms control the rNHC behavior. The present study provides new insights into the ligand design of related Ru catalysts.
The proton induced Ru-C bond variation which was previously found to be relevant in the water oxidation has been investigated using cyclometalated ruthenium complexes with three phenanthroline (phen) isomers. The designed complexes, [Ru(bpy)2(1,5-phen)]+ ([2]+), [Ru(bpy)2(1,6-phen)]+ ([3]+), and [Ru(bpy)2(1,7-phen)]+ ([4]+) were newly synthesized and their structural and electronic properties were analyzed by various spectroscopy and theoretical protocol. Protonation of [4]+ triggered profound electronic structural change to form remote N-heterocyclic carbene (rNHC), while protonation of [2]+ and [3]+ did not affect their structures. It was found that changes in the electronic structure of phen beyond classical resonance forms control the rNHC behavior. The present study provides new insights into the ligand design of related Ru catalysts. |
| 44. |
An efficient computational scheme for electronic excitation spectra of molecules in solution using the symmetry-adapted cluster-configuration interaction method: the accuracy of excitation energies and intuitive charge-transfer indices
R. Fukuda and M. Ehara J. Chem. Phys. 141, 154104-1-11 (2014) + [Abstract] 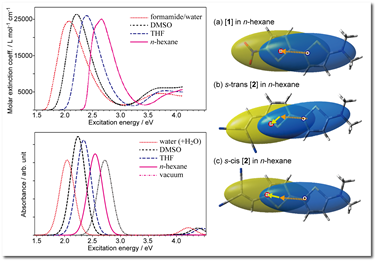 Solvent effects on electronic excitation spectra are considerable in many situations; therefore, we propose an efficient and reliable computational scheme that is based on the symmetry-adapted cluster-configuration interaction (SAC-CI) method and the polarizable continuum model (PCM) for describing electronic excitations in solution. The new scheme combines the recently proposed first-order PCM SAC-CI method with the PTE (perturbation theory at the energy level) PCM SAC scheme. This is essentially equivalent to the usual SAC and SAC-CI computations with using the PCM Hartree-Fock orbital and integrals, except for the additional correction terms that represent solute-solvent interactions. The test calculations demonstrate that the present method is a very good approximation of the more costly iterative PCM SAC-CI method for excitation energies of closed-shell molecules in their equilibrium geometry. This method provides very accurate values of electric dipole moments but is insufficient for describing the charge-transfer (CT) indices in polar solvent. The present method accurately reproduces the absorption spectra and their solvatochromism of push-pull type 2,2'-bithiophene molecules. Significant solvent and substituent effects on these molecules are intuitively visualized using the CT indices. The present method is the simplest and theoretically consistent extension of SAC-CI method for including PCM environment, and therefore, it is useful for theoretical and computational spectroscopy.
Solvent effects on electronic excitation spectra are considerable in many situations; therefore, we propose an efficient and reliable computational scheme that is based on the symmetry-adapted cluster-configuration interaction (SAC-CI) method and the polarizable continuum model (PCM) for describing electronic excitations in solution. The new scheme combines the recently proposed first-order PCM SAC-CI method with the PTE (perturbation theory at the energy level) PCM SAC scheme. This is essentially equivalent to the usual SAC and SAC-CI computations with using the PCM Hartree-Fock orbital and integrals, except for the additional correction terms that represent solute-solvent interactions. The test calculations demonstrate that the present method is a very good approximation of the more costly iterative PCM SAC-CI method for excitation energies of closed-shell molecules in their equilibrium geometry. This method provides very accurate values of electric dipole moments but is insufficient for describing the charge-transfer (CT) indices in polar solvent. The present method accurately reproduces the absorption spectra and their solvatochromism of push-pull type 2,2'-bithiophene molecules. Significant solvent and substituent effects on these molecules are intuitively visualized using the CT indices. The present method is the simplest and theoretically consistent extension of SAC-CI method for including PCM environment, and therefore, it is useful for theoretical and computational spectroscopy.
|
| 43. |
Rydberg and π-π* transitions in film surfaces of various kinds of nylons studied by attenuated total reflection-far-ultraviolet spectroscopy and quantum chemical calculations: peak shifts in the spectra and their relation to nylon structure and hydrogen bondings
Y. Morisawa, M. Yasunaga, H. Sato, R. Fukuda, M. Ehara, and Y. Ozaki J. Phys. Chem. B 118, 11855-11861 (2014) + [Abstract] 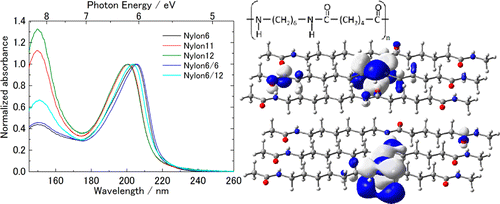 Attenuated total reflection far-ultraviolet (ATR-FUV) spectra in the 145-260 nm region were measured for surfaces (thickness: 50-200 nm) of various kinds of Nylons in cast films to explore their electronic transitions in the FUV region. ATR-FUV spectra show two major bands near 150 and 200 nm in the surface condensed phase of Nylons. Transmittance (Tr) spectra were also observed in particular for the analysis of valence excitations. Time-dependent density functional theory (TD-DFT/ CAM-B3LYP) calculations were carried out using the model systems to provide the definitive assignments of their absorption spectra and to elucidate their peak shifts in several Nylons, in particular, focusing on their crystal alignment structures and intermolecular hydrogen bondings. Two major bands of Nylon films near 150 and 200 nm are characterized as σ-Rydberg 3p and π-π* transitions of Nylons, respectively. These assignments are also coherent with those of liquid n-alkanes (n=5-14) and liquid amides observed previously. The Rydberg transitions are delocalized over the hydrocarbon chains, while the π-π* transitions are relatively localized at the amide group. Differences in the peak positions and intensity were found in both ATR- and Tr-FUV spectra for different Nylons. A redshift of the π-π* amide band in the FUV spectra of Nylon 6 and Nylon 6/6 models in α form is attributed to the crystal structure pattern and the intermolecular hydrogen bondings, which result in the different delocalization character of the π-π* transitions and transition dipole coupling.
Attenuated total reflection far-ultraviolet (ATR-FUV) spectra in the 145-260 nm region were measured for surfaces (thickness: 50-200 nm) of various kinds of Nylons in cast films to explore their electronic transitions in the FUV region. ATR-FUV spectra show two major bands near 150 and 200 nm in the surface condensed phase of Nylons. Transmittance (Tr) spectra were also observed in particular for the analysis of valence excitations. Time-dependent density functional theory (TD-DFT/ CAM-B3LYP) calculations were carried out using the model systems to provide the definitive assignments of their absorption spectra and to elucidate their peak shifts in several Nylons, in particular, focusing on their crystal alignment structures and intermolecular hydrogen bondings. Two major bands of Nylon films near 150 and 200 nm are characterized as σ-Rydberg 3p and π-π* transitions of Nylons, respectively. These assignments are also coherent with those of liquid n-alkanes (n=5-14) and liquid amides observed previously. The Rydberg transitions are delocalized over the hydrocarbon chains, while the π-π* transitions are relatively localized at the amide group. Differences in the peak positions and intensity were found in both ATR- and Tr-FUV spectra for different Nylons. A redshift of the π-π* amide band in the FUV spectra of Nylon 6 and Nylon 6/6 models in α form is attributed to the crystal structure pattern and the intermolecular hydrogen bondings, which result in the different delocalization character of the π-π* transitions and transition dipole coupling.
|
| 42. |
Effects of perturbation-selection and orbital dependence for the SAC-CI calculations in valence excitations of medium-size molecules
R. Fukuda and M. Ehara J. Comput. Chem. 35, 2163-2176 (2014) + [Abstract] 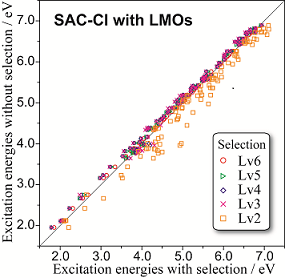 The efficiency and accuracy of the perturbation-selection used in the symmetry-adapted cluster-configuration interaction (SAC-CI) calculations are investigated for several low-lying valence excited states of 21 medium-size molecules, including typical chromophores with heterocyclic macrocycles (free-base porphine, coumarin, indole, and BODIPY), nucleobases, amino acids (tyrosine and tryptophan), polycyclic aromatic hydrocarbons, and organometallics (ferrocene and Re(bpy)(CO)4+1). Benchmark SAC-CI calculations with up to 110 million operators are performed. The efficiency of the perturbation-selection depends on the molecular orbitals (MOs); therefore, the canonical MO and localized MO (LMO) obtained by Pipek-Mezeyfs method are examined. Except for the highly symmetric molecules, using LMOs improves the efficiency and accuracy of the perturbation-selection. With using LMOs and perturbation-selection, sufficiently reliable results can be obtained in less than 10% of the computational costs required for the full-dimensional calculations. The perturbation-selection with LMOs is suggested to be a promising method for excited states in larger molecular systems.
The efficiency and accuracy of the perturbation-selection used in the symmetry-adapted cluster-configuration interaction (SAC-CI) calculations are investigated for several low-lying valence excited states of 21 medium-size molecules, including typical chromophores with heterocyclic macrocycles (free-base porphine, coumarin, indole, and BODIPY), nucleobases, amino acids (tyrosine and tryptophan), polycyclic aromatic hydrocarbons, and organometallics (ferrocene and Re(bpy)(CO)4+1). Benchmark SAC-CI calculations with up to 110 million operators are performed. The efficiency of the perturbation-selection depends on the molecular orbitals (MOs); therefore, the canonical MO and localized MO (LMO) obtained by Pipek-Mezeyfs method are examined. Except for the highly symmetric molecules, using LMOs improves the efficiency and accuracy of the perturbation-selection. With using LMOs and perturbation-selection, sufficiently reliable results can be obtained in less than 10% of the computational costs required for the full-dimensional calculations. The perturbation-selection with LMOs is suggested to be a promising method for excited states in larger molecular systems.
|
| 41. |
Benchmark Study on the Triplet Excited-State Geometries and Phosphorescence Energies of Heterocyclic Compounds: Comparison Between TD-PBE0 and SAC-CI
D. Bousquet, R. Fukuda, D. Jacquemin, I. Ciofini, C. Adamo, and M. Ehara J. Chem. Theory Comput. 10, 3969-3979 (2014) + [Abstract] 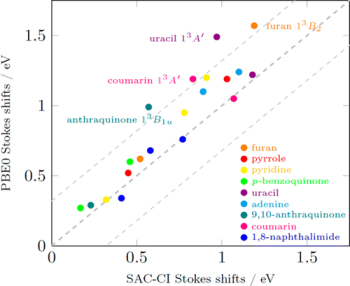 In this work, we investigated the properties of the triplet excited states of heterocyclic compounds including their geometries, electronic properties, and phosphorescence energies by using both the direct symmetry-adapted cluster-configuration interaction (SAC-CI) method and the TD-DFT approach with the PBE0 exchange-correlation functional (TD-PBE0). The target states are the ππ* and nπ* triplet states of furan, pyrrole, pyridine, p-benzoquinone, uracil, adenine, 9,10-anthraquinone, coumarin, and 1,8-naphthalimide as well as the Rydberg states. The present benchmark demonstrates that these two methods provide reasonably accurate geometries for the excited states of these heterocyclic compounds. The calculated Stokes shifts, which reflect geometry changes, were consistent for both these methods. The trends of agreement with experimental or reference values obtained for a panel of exchange-correlation functionals used to compute the absolute emission energies from the triplet states, differ from those found for the singlet excited states. Some of the low-lying triplet excited states were examined in detail for the first time, including vibrational analysis.
In this work, we investigated the properties of the triplet excited states of heterocyclic compounds including their geometries, electronic properties, and phosphorescence energies by using both the direct symmetry-adapted cluster-configuration interaction (SAC-CI) method and the TD-DFT approach with the PBE0 exchange-correlation functional (TD-PBE0). The target states are the ππ* and nπ* triplet states of furan, pyrrole, pyridine, p-benzoquinone, uracil, adenine, 9,10-anthraquinone, coumarin, and 1,8-naphthalimide as well as the Rydberg states. The present benchmark demonstrates that these two methods provide reasonably accurate geometries for the excited states of these heterocyclic compounds. The calculated Stokes shifts, which reflect geometry changes, were consistent for both these methods. The trends of agreement with experimental or reference values obtained for a panel of exchange-correlation functionals used to compute the absolute emission energies from the triplet states, differ from those found for the singlet excited states. Some of the low-lying triplet excited states were examined in detail for the first time, including vibrational analysis.
|
| 40. |
Electronic Transitions in Conformationally Controlled Tetrasilanes with a Wide Range of SiSiSiSi Dihedral Angles
H. Tsuji, H. A. Fogarty, M. Ehara, R. Fukuda, D. L. Casher, K. Tamao, H. Nakatsuji, and J. Michl Chem. Euro. J. 20, 9431-9441 (2014) + [Abstract] 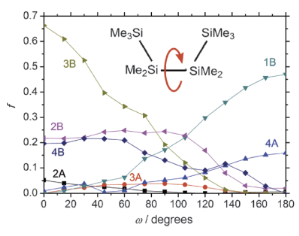 Unlike π-electron chromophores, the peralkylated n-tetrasilane
σ-electron chromophore resembles a chameleon in that its electronic spectrum changes dramatically as its silicon backbone is twisted almost effortlessly from the syn to the anti conformation (changing the SiSiSiSi dihedral angle ω from 0 to 180°). A combination of UV absorption, magnetic circular dichroism (MCD), and linear dichroism (LD) spectroscopy on conformationally controlled tetrasilanes 1-9, which cover fairly evenly the full range of angles ω, permitted a construction of an experimental correlation diagram for three to four lowest valence electronic states. The free chain tetrasilane n-Si4Me10 (10), normally present as a mixture of three enantiomeric conformer pairs of widely different angles ω, has also been included in our study. The spectral trends are interpreted in terms of avoided crossings of 1B with 2B and 2A with 3A states, in agreement with SAC-CI calculations on the excited states of 1-7 and conformers of 10.
Unlike π-electron chromophores, the peralkylated n-tetrasilane
σ-electron chromophore resembles a chameleon in that its electronic spectrum changes dramatically as its silicon backbone is twisted almost effortlessly from the syn to the anti conformation (changing the SiSiSiSi dihedral angle ω from 0 to 180°). A combination of UV absorption, magnetic circular dichroism (MCD), and linear dichroism (LD) spectroscopy on conformationally controlled tetrasilanes 1-9, which cover fairly evenly the full range of angles ω, permitted a construction of an experimental correlation diagram for three to four lowest valence electronic states. The free chain tetrasilane n-Si4Me10 (10), normally present as a mixture of three enantiomeric conformer pairs of widely different angles ω, has also been included in our study. The spectral trends are interpreted in terms of avoided crossings of 1B with 2B and 2A with 3A states, in agreement with SAC-CI calculations on the excited states of 1-7 and conformers of 10.
|
| 39. |
Cooperative H2 activation at Ag cluster/θ-Al2O3(110) dual perimeter sites: a DFT study
P. Hirunsit, K. Shimizu, R. Fukuda, S. Namuangruk, Y. Morikawa, and M. Ehara J. Phys. Chem. C 118, 7996-8006 (2014) + [Abstract] 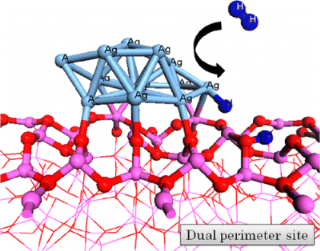 H2 dissociation by Ag clusters supported on the θ-Al2O3 (110) surface has been investigated using density functional theory calculations. The crucial role of the dual perimeter site of Ag cluster and the surface oxygen (O) site of the alumina support is demonstrated with three theoretical models: anchored cluster, isolated cluster, and anchored cluster on hydroxylated alumina. The heterolytic cleavage of H2 at the silver-alumina interface, yielding Ag-Hδ- and O-Hδ+, is thermodynamically and kinetically preferred compared with H2 cleavage at two Ag atomic sites on top of the Al2O3-supported Ag cluster and the homolytic cleavage of H2 on the isolated Ag cluster. The hydroxylation at the O site of the alumina reduces the H2 dissociation activity, which indicates that the interfacial bare O site is indispensible. It is concluded that the interfacial cooperative mechanism between the Ag cluster and Lewis acid-base pair site (bare Al-O site) is essentially relevant for the H2 activation over Ag-loaded Al2O3 catalysts.
H2 dissociation by Ag clusters supported on the θ-Al2O3 (110) surface has been investigated using density functional theory calculations. The crucial role of the dual perimeter site of Ag cluster and the surface oxygen (O) site of the alumina support is demonstrated with three theoretical models: anchored cluster, isolated cluster, and anchored cluster on hydroxylated alumina. The heterolytic cleavage of H2 at the silver-alumina interface, yielding Ag-Hδ- and O-Hδ+, is thermodynamically and kinetically preferred compared with H2 cleavage at two Ag atomic sites on top of the Al2O3-supported Ag cluster and the homolytic cleavage of H2 on the isolated Ag cluster. The hydroxylation at the O site of the alumina reduces the H2 dissociation activity, which indicates that the interfacial bare O site is indispensible. It is concluded that the interfacial cooperative mechanism between the Ag cluster and Lewis acid-base pair site (bare Al-O site) is essentially relevant for the H2 activation over Ag-loaded Al2O3 catalysts.
|
| 38. |
Electronic excitation spectra of molecules in solution calculated using the symmetry-adapted cluster-configuration interaction method in the polarizable continuum model with perturbative approach
R. Fukuda, M. Ehara, and R. Cammi J. Chem. Phys. 140, 064114-1-15 (2014) + [Abstract] 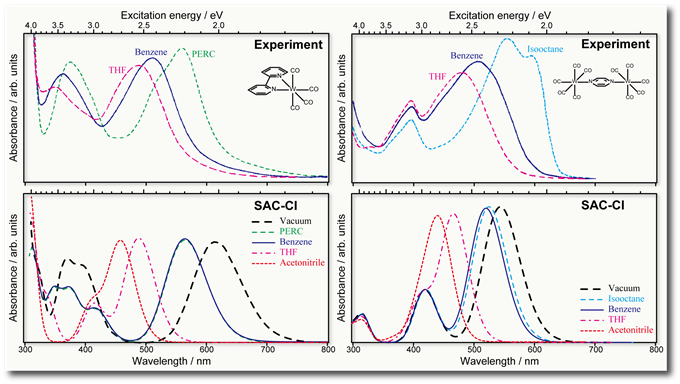 A perturbative approximation of the state specific (SS) polarizable continuum model (PCM) symmetry-adapted cluster-configuration interaction (SAC-CI) method
is proposed for efficient calculations of the electronic excitations and absorption spectra of molecules in solutions.
This first-order PCM SAC-CI method considers the solvent effects on the energies of excited states up to the first-order with using the zeroth-order wavefunctions.
This method can avoid the costly iterative procedure of the self-consistent reaction field (SCRF) calculations.
The first-order PCM SAC-CI calculations well reproduce the results obtained by the iterative method for various types of excitations of molecules in polar and nonpolar solvents.
The first-order contribution is significant for the excitation energies.
The results obtained by the zeroth-order PCM SAC-CI, which considers the fixed ground-state reaction field for the excited-state calculations, are deviated from the results
by the iterative method about 0.1 eV, and the zeroth-order PCM SAC-CI cannot predict even the direction of solvent shifts in n-hexane for many cases.
The first-order PCM SAC-CI is applied to studying the solvatochromisms of (2,2'-bipyridine)tetracarbonyltungsten [W(CO)4(bpy), bpy = 2,2'-bipyridine]
and bis(pentacarbonyltungsten)pyrazine [(OC)5W(pyz)W(CO)5, pyz = pyrazine].
The SAC-CI calculations reveal the detailed character of the excited states and the mechanisms of solvent shifts.
The energies of metal to ligand charge transfer (MLCT) states are significantly sensitive to solvents.
The first-order PCM SAC-CI well reproduces the observed absorption spectra of the tungsten carbonyl complexes in several solvents.
A perturbative approximation of the state specific (SS) polarizable continuum model (PCM) symmetry-adapted cluster-configuration interaction (SAC-CI) method
is proposed for efficient calculations of the electronic excitations and absorption spectra of molecules in solutions.
This first-order PCM SAC-CI method considers the solvent effects on the energies of excited states up to the first-order with using the zeroth-order wavefunctions.
This method can avoid the costly iterative procedure of the self-consistent reaction field (SCRF) calculations.
The first-order PCM SAC-CI calculations well reproduce the results obtained by the iterative method for various types of excitations of molecules in polar and nonpolar solvents.
The first-order contribution is significant for the excitation energies.
The results obtained by the zeroth-order PCM SAC-CI, which considers the fixed ground-state reaction field for the excited-state calculations, are deviated from the results
by the iterative method about 0.1 eV, and the zeroth-order PCM SAC-CI cannot predict even the direction of solvent shifts in n-hexane for many cases.
The first-order PCM SAC-CI is applied to studying the solvatochromisms of (2,2'-bipyridine)tetracarbonyltungsten [W(CO)4(bpy), bpy = 2,2'-bipyridine]
and bis(pentacarbonyltungsten)pyrazine [(OC)5W(pyz)W(CO)5, pyz = pyrazine].
The SAC-CI calculations reveal the detailed character of the excited states and the mechanisms of solvent shifts.
The energies of metal to ligand charge transfer (MLCT) states are significantly sensitive to solvents.
The first-order PCM SAC-CI well reproduces the observed absorption spectra of the tungsten carbonyl complexes in several solvents.
|
| 37. |
Electronic transitions in liquid amides studied by using attenuated total reflection far-ultraviolet spectroscopy and quantum chemical calculations
Y. Morisawa, M. Yasunaga, R. Fukuda, M. Ehara, and Y. Ozaki J. Chem. Phys. 139, 154301-1-9 (2013) + [Abstract] 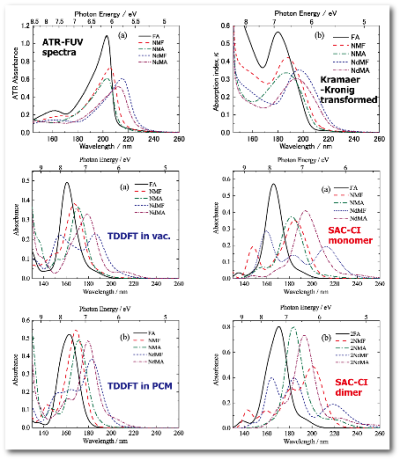 Attenuated total reflection far-ultraviolet (ATR-FUV) spectra in the 140-260 nm region were measured for several types of liquid amides
(formamide, FA; N-N-methylformamide, NMF; N-methylacetamide, NMA; N,N-dimethylformamide, NdMF; and N,N-dimethylacetamide, NdMA)
to investigate their electronic transitions in the FUV region. The spectra were compared with the corresponding gas-phase
spectra to examine the shift in the major absorption band in the 180-200 nm region going from the gas phase to the liquid phase,
and it was found that the peak shift was dependent on the particular amide.
FA and NMF, which exhibit intermolecular C=O···H-N hydrogen bonding, show a large shift of ~0.60 eV to lower energy;
however, NMA, which also exhibits hydrogen bonding, shows only a small shift.
In NdMF and NdMA, C=O groups seem to be coupled, which results in a small peak shift.
Two types of quantum chemical calculations, time-dependent density functional theory (TD-DFT) and
symmetry-adapted-cluster configuration interaction (SAC-CI) method, were performed to elucidate the origin of the shifts and the band assignments.
The shift estimated by the monomer and dimer models with TD-DFT reproduced well the observed shift from the gas phase to the liquid phase.
This suggests that the intermolecular hydrogen-bonding interaction significantly affects the magnitude of the shift.
The many-body effects were also considered using the larger cluster models (trimer to pentamer).
The energy shift calculated using SAC-CI with the monomer and the state-specific polarizable continuum model was also accurate,
indicating that the nonlinear polarization effect appears to be important. As for the band assignments,
it was found that though the major band can be mainly attributed to the π-π* transition, several types of Rydberg transitions also
exist in its vicinity and mixing of orbitals with the same symmetry occurs.
The number and type of Rydberg transitions in the spectra depend upon the type of amide molecules.
The valence-Rydberg coupling of the π-π* transition is more significant than n-π* transition, which also holds in the pure liquid phase.
Attenuated total reflection far-ultraviolet (ATR-FUV) spectra in the 140-260 nm region were measured for several types of liquid amides
(formamide, FA; N-N-methylformamide, NMF; N-methylacetamide, NMA; N,N-dimethylformamide, NdMF; and N,N-dimethylacetamide, NdMA)
to investigate their electronic transitions in the FUV region. The spectra were compared with the corresponding gas-phase
spectra to examine the shift in the major absorption band in the 180-200 nm region going from the gas phase to the liquid phase,
and it was found that the peak shift was dependent on the particular amide.
FA and NMF, which exhibit intermolecular C=O···H-N hydrogen bonding, show a large shift of ~0.60 eV to lower energy;
however, NMA, which also exhibits hydrogen bonding, shows only a small shift.
In NdMF and NdMA, C=O groups seem to be coupled, which results in a small peak shift.
Two types of quantum chemical calculations, time-dependent density functional theory (TD-DFT) and
symmetry-adapted-cluster configuration interaction (SAC-CI) method, were performed to elucidate the origin of the shifts and the band assignments.
The shift estimated by the monomer and dimer models with TD-DFT reproduced well the observed shift from the gas phase to the liquid phase.
This suggests that the intermolecular hydrogen-bonding interaction significantly affects the magnitude of the shift.
The many-body effects were also considered using the larger cluster models (trimer to pentamer).
The energy shift calculated using SAC-CI with the monomer and the state-specific polarizable continuum model was also accurate,
indicating that the nonlinear polarization effect appears to be important. As for the band assignments,
it was found that though the major band can be mainly attributed to the π-π* transition, several types of Rydberg transitions also
exist in its vicinity and mixing of orbitals with the same symmetry occurs.
The number and type of Rydberg transitions in the spectra depend upon the type of amide molecules.
The valence-Rydberg coupling of the π-π* transition is more significant than n-π* transition, which also holds in the pure liquid phase.
|
| 36. |
Intuitive indexes for charge-transfer excitation based on SAC-CI and TD-DFT calculations
M. Ehara, R. Fukuda, C. Adamo, and I. Ciofini J. Comput. Chem. 34, 2498-2501 (2013) + [Abstract] 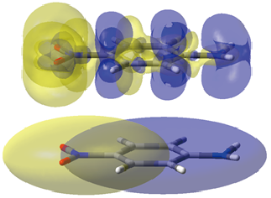 A recently proposed charge-transfer (CT) index and some related quantities aimed to the description of
CT excitations in push-pull donor-acceptor model systems were computed in vacuum and in ethanol by
the direct SAC-CI method including solvent effects by means of the nonequilibrium state-specific approach.
The effects of both solvation and electron correlations on these quantities were found to be significant.
The present results are also in line with previous TD-DFT calculations and they demonstrate that SAC-CI provides
a description of the excitation character close to that of TD-PBE0. Indeed, CT indices evaluated by
the SAC-CI and TD-PBE0 would be useful in the field of materials chemistry,
for the design and development of novel molecular materials.
A recently proposed charge-transfer (CT) index and some related quantities aimed to the description of
CT excitations in push-pull donor-acceptor model systems were computed in vacuum and in ethanol by
the direct SAC-CI method including solvent effects by means of the nonequilibrium state-specific approach.
The effects of both solvation and electron correlations on these quantities were found to be significant.
The present results are also in line with previous TD-DFT calculations and they demonstrate that SAC-CI provides
a description of the excitation character close to that of TD-PBE0. Indeed, CT indices evaluated by
the SAC-CI and TD-PBE0 would be useful in the field of materials chemistry,
for the design and development of novel molecular materials.
|
| 35. |
Electronic excited states and electronic spectra of biphenyl: a study using many-body wavefunction methods and density functional theories
R. Fukuda and M. Ehara Phys. Chem. Chem. Phys. 15, 17426-17434 (2013) + [Abstract] 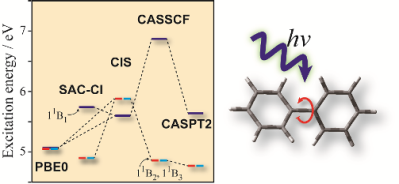 The low-lying electronic excited states of biphenyl were studied with the symmetry-adapted cluster-configuration interaction (SAC-CI),
complete active space self-consistent field (CASSCF), complete active space perturbation theory of the second-order (CASPT2),
and the time-dependent density functional theory (TDDFT).
The molecular geometries in the ground and excited states were optimized with using the SAC-CI and TDDFT for singlet and triplet states.
The energies of vertical excitations, emissions, and adiabatic transitions were calculated.
The TDDFT calculations significantly underestimated the excitation energy of the 11B1 state, while the SAC-CI and CASPT2 provided the essentially similar results.
The present SAC-CI and CASPT2 calculations concluded that the lowest singlet state of isolated biphenyl is the 11B3 state
that takes a planar geometry and the second lowest state is the 11B2 state with a twisted geometry.
The present results were consistent with the previous experimental findings.
The 11B1 state that has a charge-separated biracial character in the vertical excitation relaxed into a planar quinoide structure
in which bond alternations were emphasized. The other states took a benzenoid structure.
The ultraviolet (UV) absorption and circular dichroism (CD) spectra below 7 eV were calculated with the SAC-CI method.
The valence-Rydberg mixings were found to be significant in the second and higher series of excited states.
The low-lying electronic excited states of biphenyl were studied with the symmetry-adapted cluster-configuration interaction (SAC-CI),
complete active space self-consistent field (CASSCF), complete active space perturbation theory of the second-order (CASPT2),
and the time-dependent density functional theory (TDDFT).
The molecular geometries in the ground and excited states were optimized with using the SAC-CI and TDDFT for singlet and triplet states.
The energies of vertical excitations, emissions, and adiabatic transitions were calculated.
The TDDFT calculations significantly underestimated the excitation energy of the 11B1 state, while the SAC-CI and CASPT2 provided the essentially similar results.
The present SAC-CI and CASPT2 calculations concluded that the lowest singlet state of isolated biphenyl is the 11B3 state
that takes a planar geometry and the second lowest state is the 11B2 state with a twisted geometry.
The present results were consistent with the previous experimental findings.
The 11B1 state that has a charge-separated biracial character in the vertical excitation relaxed into a planar quinoide structure
in which bond alternations were emphasized. The other states took a benzenoid structure.
The ultraviolet (UV) absorption and circular dichroism (CD) spectra below 7 eV were calculated with the SAC-CI method.
The valence-Rydberg mixings were found to be significant in the second and higher series of excited states.
|
| 34. |
Theoretical study of the electronic excitations of free-base porphyrin-Ar2 van der Waals complexes R. Fukuda and M. Ehara J. Chem. Phys. 139, 074303-1-10 (2013) + [Abstract] 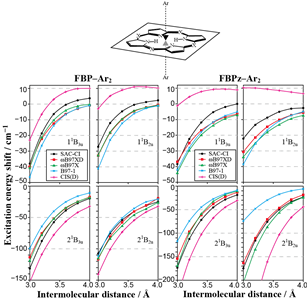 The intermolecular interaction of free-base porphine (FBP)-Ar2 and free-base tetraazaporphyrin (FBPz)-Ar2 van der Waals (vdW) complexes
was calculated in the ground state and vertical excitations that correspond to the Q- and B-bands using the many-body wavefunction theory of
the symmetry-adapted cluster-configuration interaction (SAC-CI) method and time-dependent density functional theory (TDDFT).
For the 11B3u state of FBP-Ar2 a blueshift (high-energy shift) of excitation energy was calculated using the SAC-CI method;
such a blueshift was not obtained by TDDFT calculations.
This calculated blueshift corresponded to the experimentally observed blueshift in the Qx-band of FBP for FBP-Arn complexes.
For FBPz-Ar2, blueshifts of the Q-band were not obtained using SAC-CI and TDDFT.
These behaviors of the energy shift of the Q-bands could not be explained by the point dipole-point dipole interaction model. Large redshifts (low-energy shift)
were obtained for the B-band states (21B3u and 21B2u) of FBP and FBPz.
The energy shift showed the inverse sixth-power dependence on the intermolecular distance.
The point dipole-point dipole interaction model can describe the redshift of the B-band.
For the excited states that exhibit large redshifts, the TDDFT can qualitatively describe the vdW interaction in the excited states by supermolecular calculations.
The solvatochromic shifts for FBP and FBPz in an Ar matrix were examined by the linear-response polarizable continuum model and TDDFT.
The magnitude of calculated solvatochromic redshifts was proportional to the square of the transition dipole moment.
The intermolecular interaction of free-base porphine (FBP)-Ar2 and free-base tetraazaporphyrin (FBPz)-Ar2 van der Waals (vdW) complexes
was calculated in the ground state and vertical excitations that correspond to the Q- and B-bands using the many-body wavefunction theory of
the symmetry-adapted cluster-configuration interaction (SAC-CI) method and time-dependent density functional theory (TDDFT).
For the 11B3u state of FBP-Ar2 a blueshift (high-energy shift) of excitation energy was calculated using the SAC-CI method;
such a blueshift was not obtained by TDDFT calculations.
This calculated blueshift corresponded to the experimentally observed blueshift in the Qx-band of FBP for FBP-Arn complexes.
For FBPz-Ar2, blueshifts of the Q-band were not obtained using SAC-CI and TDDFT.
These behaviors of the energy shift of the Q-bands could not be explained by the point dipole-point dipole interaction model. Large redshifts (low-energy shift)
were obtained for the B-band states (21B3u and 21B2u) of FBP and FBPz.
The energy shift showed the inverse sixth-power dependence on the intermolecular distance.
The point dipole-point dipole interaction model can describe the redshift of the B-band.
For the excited states that exhibit large redshifts, the TDDFT can qualitatively describe the vdW interaction in the excited states by supermolecular calculations.
The solvatochromic shifts for FBP and FBPz in an Ar matrix were examined by the linear-response polarizable continuum model and TDDFT.
The magnitude of calculated solvatochromic redshifts was proportional to the square of the transition dipole moment.
|
| 33. |
Excited-state geometries of heteroaromatic compounds: a comparative TD-DFT and SAC-CI study
D. Bousquet, R. Fukuda, P. Maitarad, D. Jacquemin, I. Ciofini, C. Adamo, and M. Ehara J. Chem. Theory Comput. 9, 2368-2379 (2013) + [Abstract] 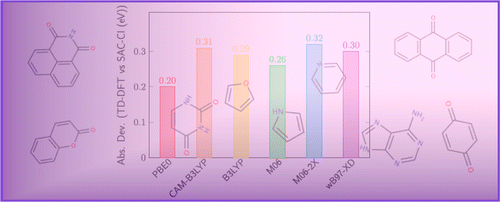 The structures of low-lying singlet excited states of nine π-conjugated heteroaromatic compounds
have been investigated by the symmetry-adapted cluster-configuration interaction (SAC-CI) method
and the time-dependent density functional theory (TDDFT) using the PBE0 functional (TD-PBE0).
In particular, the geometry relaxation in some ππ* and nπ* excited states of furan, pyrrole, pyridine,
p-benzoquinone, uracil, adenine, 9,10-anthraquinone, coumarin, and 1,8-naphthalimide as well as
the corresponding vertical transitions, including Rydberg excited states, have been analyzed in details.
The basis set and functional dependence of the results was also examined. The SAC-CI and TD-PBE0 calculations
showedreasonable agreement in both transition energies and excited-state equilibrium structures for these heteroaromatic compounds.
The structures of low-lying singlet excited states of nine π-conjugated heteroaromatic compounds
have been investigated by the symmetry-adapted cluster-configuration interaction (SAC-CI) method
and the time-dependent density functional theory (TDDFT) using the PBE0 functional (TD-PBE0).
In particular, the geometry relaxation in some ππ* and nπ* excited states of furan, pyrrole, pyridine,
p-benzoquinone, uracil, adenine, 9,10-anthraquinone, coumarin, and 1,8-naphthalimide as well as
the corresponding vertical transitions, including Rydberg excited states, have been analyzed in details.
The basis set and functional dependence of the results was also examined. The SAC-CI and TD-PBE0 calculations
showedreasonable agreement in both transition energies and excited-state equilibrium structures for these heteroaromatic compounds.
|
| 32. |
Theoretical study on the excited electronic states of coronene and its π-extended molecules using the symmetry-adapted cluster-configuration interaction method
R. Fukuda and M. Ehara Bull. Chem. Soc. Jpn. 86, 445-451 (2013) + [Abstract] 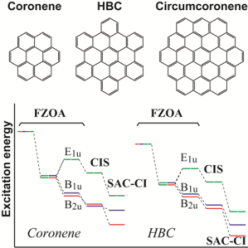 The excited electronic states and optical absorption spectra of coronene (C24H12),
hexa-peri-hexabenzocoronene (HBC) (C42H18),
and circumcoronene (C54H18) were studied using
the symmetry-adapted cluster-configuration interaction (SAC-CI) method.
For coronene and HBC, the SAC-CI calculations reproduced the experimental spectra well and predicted optically forbidden excited states.
For HBC, the symmetry lowering enhanced the intensity of the S2 state that corresponds to the p-band,
and the SAC-CI calculation predicted the existence of the second and third optically-allowed states around the β-band region near 4.0 eV.
For circumcoronene, the SAC-CI calculation predicted a strong absorption of the β-band in the visible light region.
The mechanisms of energy splitting for the HOMO?LUMO transition were investigated.
Electron correlation was the most important factor for the energy splitting between the lowest and the next-lowest states.
Configuration interaction with single excitations (CIS) calculations could not correctly predict the relative energies of these states in coronene and circumcoronene.
For HBC, on the other hand, the CIS calculation provided the same energy order as the SAC-CI calculation.
The excited electronic states and optical absorption spectra of coronene (C24H12),
hexa-peri-hexabenzocoronene (HBC) (C42H18),
and circumcoronene (C54H18) were studied using
the symmetry-adapted cluster-configuration interaction (SAC-CI) method.
For coronene and HBC, the SAC-CI calculations reproduced the experimental spectra well and predicted optically forbidden excited states.
For HBC, the symmetry lowering enhanced the intensity of the S2 state that corresponds to the p-band,
and the SAC-CI calculation predicted the existence of the second and third optically-allowed states around the β-band region near 4.0 eV.
For circumcoronene, the SAC-CI calculation predicted a strong absorption of the β-band in the visible light region.
The mechanisms of energy splitting for the HOMO?LUMO transition were investigated.
Electron correlation was the most important factor for the energy splitting between the lowest and the next-lowest states.
Configuration interaction with single excitations (CIS) calculations could not correctly predict the relative energies of these states in coronene and circumcoronene.
For HBC, on the other hand, the CIS calculation provided the same energy order as the SAC-CI calculation.
|
| 31. |
Mechanism of aerobic oxidation of methanol to formic acid on Au8-: a DFT study
S. Karanjit, K. Bobuatong, R. Fukuda, M. Ehara, and H. Sakurai Int. J. Quantum Chem. 113, 428-436 (2013) + [Abstract] 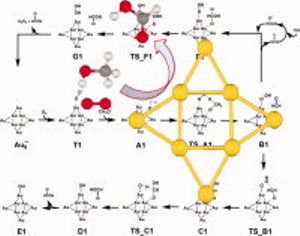 The mechanism of the aerobic oxidation of methanol to formic acid catalyzed by Au8- has been systematically investigated using density functional theory with the M06 functional.
The reaction pathways were examined by taking into account the full structural relaxation of the Au8-.
Stepwise and concerted reaction mechanisms are proposed.
The stepwise mechanism is initiated by the hydrogen abstraction of a methoxy species by a superoxo-like anion on the gold cluster, resulting in the formation of formaldehyde.
Subsequently, the formaldehyde is activated by the hydroxyl group of a hydroperoxyl-like complex, leading to the formation of a hemiacetal intermediate.
The formation of formic acid in the final step is achieved by hydrogen abstraction of the hemiacetal intermediate by atomic oxygen attached to the gold cluster.
Our calculations indicate that the first step of the stepwise mechanism, that is, hydrogen abstraction of the methoxy species, is the rate-determining step.
Another possible reaction pathway involving a single-step hydrogen abstraction, a concerted mechanism, is also discussed.
This mechanism may also be responsible for the reasonable catalytic activity of aerobic oxidation of methanol on Au8- because of the low activation energy barrier.
The mechanism of the aerobic oxidation of methanol to formic acid catalyzed by Au8- has been systematically investigated using density functional theory with the M06 functional.
The reaction pathways were examined by taking into account the full structural relaxation of the Au8-.
Stepwise and concerted reaction mechanisms are proposed.
The stepwise mechanism is initiated by the hydrogen abstraction of a methoxy species by a superoxo-like anion on the gold cluster, resulting in the formation of formaldehyde.
Subsequently, the formaldehyde is activated by the hydroxyl group of a hydroperoxyl-like complex, leading to the formation of a hemiacetal intermediate.
The formation of formic acid in the final step is achieved by hydrogen abstraction of the hemiacetal intermediate by atomic oxygen attached to the gold cluster.
Our calculations indicate that the first step of the stepwise mechanism, that is, hydrogen abstraction of the methoxy species, is the rate-determining step.
Another possible reaction pathway involving a single-step hydrogen abstraction, a concerted mechanism, is also discussed.
This mechanism may also be responsible for the reasonable catalytic activity of aerobic oxidation of methanol on Au8- because of the low activation energy barrier.
|
| 30. |
Mechanisms for solvatochromic shifts of free-base porphine studied with polarizable continuum models and explicit solute-solvent interactions
R. Fukuda and M. Ehara J. Chem. Theory Comput. 9, 470-480 (2013) + [Abstract] 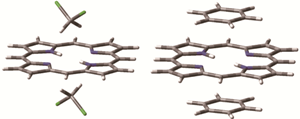 Solvatochromic shifts of free-base porphine in the Q-band and B-band were studied using the polarizable continuum model (PCM) and explicit solvent molecules
employing time-dependent density functional theory (TDDFT) and the symmetry-adapted cluster-configuration interaction (SAC-CI) method.
The state-specific (SS) and linear-response (LR) methods were examined in the PCM calculations.
These models involve different types of solute-solvent interactions.
The LR PCM and explicit solvation models reproduced the experimentally observed trends of the solvatochromic shifts,
while the SS PCM failed to reproduce the experimental findings.
The origin of the solvatochromic shifts of free-base porphine was dispersive interactions between the solute and solvent.
Specific solute-solvent interactions would be important for a decrease of the splitting width between Q-bands.
Based on the Casimir-Polder formula and a decomposition analysis, it was found that the dominant part of the solute-solvent interactions
can be considered using independent particle approximations.
Solvatochromic shifts of free-base porphine in the Q-band and B-band were studied using the polarizable continuum model (PCM) and explicit solvent molecules
employing time-dependent density functional theory (TDDFT) and the symmetry-adapted cluster-configuration interaction (SAC-CI) method.
The state-specific (SS) and linear-response (LR) methods were examined in the PCM calculations.
These models involve different types of solute-solvent interactions.
The LR PCM and explicit solvation models reproduced the experimentally observed trends of the solvatochromic shifts,
while the SS PCM failed to reproduce the experimental findings.
The origin of the solvatochromic shifts of free-base porphine was dispersive interactions between the solute and solvent.
Specific solute-solvent interactions would be important for a decrease of the splitting width between Q-bands.
Based on the Casimir-Polder formula and a decomposition analysis, it was found that the dominant part of the solute-solvent interactions
can be considered using independent particle approximations.
|
| 29. |
D-D-π-A type organic dyes for dye-sensitized solar cells with a potential of direct electron injection
and a high extinction coefficient: synthesis, characterization, and theoretical investigation
S. Namuangruk, R. Fukuda, M. Ehara, J. Meeprasert, T. Khanasa, S. Morada, T. Kaewin, S. Jungsuttiwong, T. Sudyoadsuk, and V. Promarak J. Phys. Chem. C 116, 25653-25663 (2012) + [Abstract] 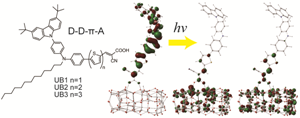 A series of organic sensitizers with the direct electron injection mechanism and high molar extinction coefficient comprising double donors,
π-spacer, and anchoring acceptor groups (D-D-π-A type) were synthesized and characterized by experimental and theoretical methods for dye-sensitized solar cells.
(E)-2-cyano-3-(5"-(4-((4-(3,6-di- tert-butylcarbazol-9-yl)phe-nyl)(dodecyl)amino)phenyl) -[2,2':5',2"-terthiophen]-5-yl) acrylic acid (UB3) showed the performance with
a maximal IPCE value of 83%, Jsc value of 10.89 mA cm-2, Voc value of 0.70 V, and FF value of 0.67,
that correspond to an overall conversion efficiency of 5.12% under AM 1.5G illumination.
The molecular geometry, electronic structure, and excited states were investigated with the density functional theory,
time-dependent density functional theory, and symmetry-adapted cluster-configuration interaction method.
The double donor moieties are contributed not only to enhance the electron-donating ability,
but also inhibit aggregation between dye molecules and prevent iodide/triiodide in the electrolyte from recombining with injected electrons in TiO2.
Detailed assignments of the UV-visible spectra below ionization threshold were given.
The low-lying light harvesting state has intramolecular charge transfer character with high molar extinction coefficient because of the long π-spacer.
Our experimental and theoretical findings supported the potential of direct electron injection from dye to TiO2 by one-step with electronic excitation for
the present D-D-π-A sensitizers. The direct electron injection, inhibited aggregation, and high molar extinction coefficient may be the origin of the observed high efficiency.
This type of D-D-π-A structure with direct electron injection would simplify the strategy for designing organic sensitizers.
A series of organic sensitizers with the direct electron injection mechanism and high molar extinction coefficient comprising double donors,
π-spacer, and anchoring acceptor groups (D-D-π-A type) were synthesized and characterized by experimental and theoretical methods for dye-sensitized solar cells.
(E)-2-cyano-3-(5"-(4-((4-(3,6-di- tert-butylcarbazol-9-yl)phe-nyl)(dodecyl)amino)phenyl) -[2,2':5',2"-terthiophen]-5-yl) acrylic acid (UB3) showed the performance with
a maximal IPCE value of 83%, Jsc value of 10.89 mA cm-2, Voc value of 0.70 V, and FF value of 0.67,
that correspond to an overall conversion efficiency of 5.12% under AM 1.5G illumination.
The molecular geometry, electronic structure, and excited states were investigated with the density functional theory,
time-dependent density functional theory, and symmetry-adapted cluster-configuration interaction method.
The double donor moieties are contributed not only to enhance the electron-donating ability,
but also inhibit aggregation between dye molecules and prevent iodide/triiodide in the electrolyte from recombining with injected electrons in TiO2.
Detailed assignments of the UV-visible spectra below ionization threshold were given.
The low-lying light harvesting state has intramolecular charge transfer character with high molar extinction coefficient because of the long π-spacer.
Our experimental and theoretical findings supported the potential of direct electron injection from dye to TiO2 by one-step with electronic excitation for
the present D-D-π-A sensitizers. The direct electron injection, inhibited aggregation, and high molar extinction coefficient may be the origin of the observed high efficiency.
This type of D-D-π-A structure with direct electron injection would simplify the strategy for designing organic sensitizers.
|
| 28. |
Optical absorption and fluorescence of PRODAN in solution:
Quantum chemical study based on the symmetry-adapted cluster-configuration interaction method
R. Fukuda, R. Chidthong, R. Cammi, and M. Ehara Chem. Phys. Lett. 552, 53-57 (2012) + [Abstract] 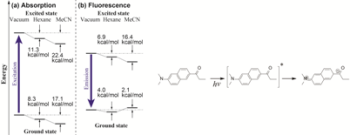 The optical absorption and fluorescence properties of 6-propionyl-2-(N,N-dimethyl)aminonaphthalene (PRODAN),
a biological probe, were investigated using the symmetry-adapted cluster-configuration interaction (SAC-CI) method.
The solvent effects were included using the polarizable continuum model (PCM).
The PCM SAC/SAC-CI calculations well reproduced the significant solvatochromic shifts, without assuming twisted charge-transfer conformations.
The geometry relaxation in naphthalene ring was significant for the lowest excited state.
The nonequilibrium solvation was important in explaining the large solvatochromic shifts of the fluorescence.
The present SAC-CI calculation showed that the absorption maximum corresponds to the second π-π* state, not the lowest excited state.
The optical absorption and fluorescence properties of 6-propionyl-2-(N,N-dimethyl)aminonaphthalene (PRODAN),
a biological probe, were investigated using the symmetry-adapted cluster-configuration interaction (SAC-CI) method.
The solvent effects were included using the polarizable continuum model (PCM).
The PCM SAC/SAC-CI calculations well reproduced the significant solvatochromic shifts, without assuming twisted charge-transfer conformations.
The geometry relaxation in naphthalene ring was significant for the lowest excited state.
The nonequilibrium solvation was important in explaining the large solvatochromic shifts of the fluorescence.
The present SAC-CI calculation showed that the absorption maximum corresponds to the second π-π* state, not the lowest excited state.
|
| 27. |
Electronic excitations of C60 fullerene calculated using the ab initio cluster expansion method
R. Fukuda and M. Ehara J. Chem. Phys. 137, 134304-1-7 (2012) + [Abstract] 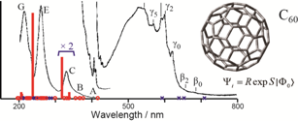 The electronic excited states and optical absorption spectrum of C60 fullerene below 6.2 eV (200 nm) were calculated using
the ab initio many-body wavefunction theory of cluster expansion method: the symmetry-adapted cluster-configuration interaction method.
Not only optically allowed states but also optically forbidden states were calculated for studying the observed weak absorptions in the visible region.
The lowest calculated singlet excited state was the 11Gg state.
The electron correlation effects are important in determining the energy levels of the four low-lying states that have the character of degenerated HOMO-LUMO transition.
The lowest optically allowed 11T1u state was calculated at 3.67 eV;
this is significantly higher than the energy values found in previous density functional calculations.
The observed weak absorption around 3.08 eV appears to correspond to the optically forbidden 11T2u state with intensity borrowing via vibronic couplings.
The electronic excited states and optical absorption spectrum of C60 fullerene below 6.2 eV (200 nm) were calculated using
the ab initio many-body wavefunction theory of cluster expansion method: the symmetry-adapted cluster-configuration interaction method.
Not only optically allowed states but also optically forbidden states were calculated for studying the observed weak absorptions in the visible region.
The lowest calculated singlet excited state was the 11Gg state.
The electron correlation effects are important in determining the energy levels of the four low-lying states that have the character of degenerated HOMO-LUMO transition.
The lowest optically allowed 11T1u state was calculated at 3.67 eV;
this is significantly higher than the energy values found in previous density functional calculations.
The observed weak absorption around 3.08 eV appears to correspond to the optically forbidden 11T2u state with intensity borrowing via vibronic couplings.
|
| 26. |
A Comparative study of C^N and N^C type cyclometalated ruthenium complexes with an NAD+/NADH function
S. K. Padhi, R. Fukuda, M. Ehara, and K. Tanaka Inorg. Chem. 51, 8091-8102 (2012) + [Abstract] 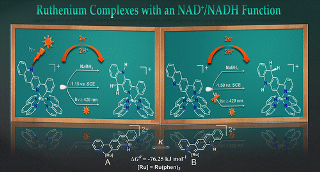 Cyclometalated ruthenium complexes having C^N and N^C type coordinating ligands with NAD+/NADH function have been synthesized and characterized by spectroscopic methods.
The variation of the coordinating position of σ-donating carbon atom leads to a drastic change in their properties.
Both the complex Ru(phbn)(phen)2]PF6( [1] PF6) and
[Ru(pad)(phen)2]PF6([2]PF6) reduced to
Ru(phbnHH)(phen)2]PF6([1HH]PF6) and
[Ru(padHH)(phen)2]PF6([2HH]PF6) by chemical and electrochemical methods.
Complex [1]PF6 photochemically reduced to [1HH]PF6 in the presence of the sacrificial agent triethylamine (TEA)
upon irradiation of visible light (λ ≥ 420 nm), whereas photochemical reduction of [2]PF6 was not successful.
Both experimental results and theoretical calculations reveal that upon protonation the energy level of the π* orbital of either of
the ligands phbn or pad is drastically stabilized compared to the nonprotonated forms.
In the protonated complex [Ru(padH)(phen)2](PF6)2{[2H](PF6)2},
the Ru-C bond exists in a tautomeric equilibrium with Ru=C coordination and behaves as a remote N-heterocyclic carbene (rNHC) complex;
on the contrary, this behavior could not be observed in protonated complex
[Ru(phbnH)(phen)2](PF6)2{[1H](PF6) 2}.
Cyclometalated ruthenium complexes having C^N and N^C type coordinating ligands with NAD+/NADH function have been synthesized and characterized by spectroscopic methods.
The variation of the coordinating position of σ-donating carbon atom leads to a drastic change in their properties.
Both the complex Ru(phbn)(phen)2]PF6( [1] PF6) and
[Ru(pad)(phen)2]PF6([2]PF6) reduced to
Ru(phbnHH)(phen)2]PF6([1HH]PF6) and
[Ru(padHH)(phen)2]PF6([2HH]PF6) by chemical and electrochemical methods.
Complex [1]PF6 photochemically reduced to [1HH]PF6 in the presence of the sacrificial agent triethylamine (TEA)
upon irradiation of visible light (λ ≥ 420 nm), whereas photochemical reduction of [2]PF6 was not successful.
Both experimental results and theoretical calculations reveal that upon protonation the energy level of the π* orbital of either of
the ligands phbn or pad is drastically stabilized compared to the nonprotonated forms.
In the protonated complex [Ru(padH)(phen)2](PF6)2{[2H](PF6)2},
the Ru-C bond exists in a tautomeric equilibrium with Ru=C coordination and behaves as a remote N-heterocyclic carbene (rNHC) complex;
on the contrary, this behavior could not be observed in protonated complex
[Ru(phbnH)(phen)2](PF6)2{[1H](PF6) 2}.
|
| 25. |
Photo-isomerization and proton-coupled electron transfer (PCET) promoted water oxidation by mononuclear cyclometalated ruthenium catalysts
S. K. Padhi, R. Fukuda, M. Ehara, and K. Tanaka Inorg. Chem. 51, 5386-5392 (2012) + [Abstract] 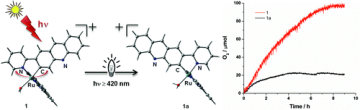 Photoisomeric transformations in ruthenium polypyridyl complexes have been rarely reported.
Herein we report the geometrical transformation of cyclometalated trans-[Ru(tpy)(PAD)(OH2)]+ ([1]+) to
the cis-[Ru(tpy)(PAD)(OH2)]+ ([1a]+) (tpy = 2,2';6',2"-terpyridine, PAD = 2-(pyrid-2'-yl)acridine)
isomer upon irradiation of visible light (λ ≥ 420 nm).
Due to a proton-induced tautomeric equilibrium between the Ru-C bond and Ru=C coordination,
the π* energy levels of PADH are lower than those of tpy by 12.61 and 12.24 kcal mol-1, respectively,
in [1]+ and [1a]+.
Isomers [1]+ and [1a]+ both act as catalytic oxygen-evolving complexes (OECs) chemically as well as electrochemically.
Photoisomeric transformations in ruthenium polypyridyl complexes have been rarely reported.
Herein we report the geometrical transformation of cyclometalated trans-[Ru(tpy)(PAD)(OH2)]+ ([1]+) to
the cis-[Ru(tpy)(PAD)(OH2)]+ ([1a]+) (tpy = 2,2';6',2"-terpyridine, PAD = 2-(pyrid-2'-yl)acridine)
isomer upon irradiation of visible light (λ ≥ 420 nm).
Due to a proton-induced tautomeric equilibrium between the Ru-C bond and Ru=C coordination,
the π* energy levels of PADH are lower than those of tpy by 12.61 and 12.24 kcal mol-1, respectively,
in [1]+ and [1a]+.
Isomers [1]+ and [1a]+ both act as catalytic oxygen-evolving complexes (OECs) chemically as well as electrochemically.
|
| 24. |
Excited states and electronic spectra of annulated dinuclear free-base phthalocyanines: a theoretical study on near-IR-absorbing dyes
R. Fukuda and M. Ehara J. Chem. Phys. 136, 114304-1--15 (2012) + [Abstract] 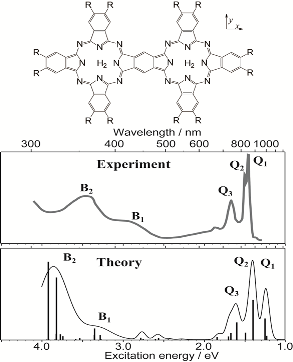 The electronic excited states and electronic absorption spectra of annulated dinuclear free-base phthalocyanine (C58H30N16)
are studied through quantum chemical calculations using the symmetryadapted cluster-configuration interaction (SAC-CI) method.
Three tautomers are possible with respect to the position of the pyrrole protons; therefore, the SAC-CI calculations for these tautomers were performed.
The structures of the Q-band states are discussed based on the character of their molecular orbitals.
The lower energy shift of the Q-bands because of dimerization is explained by the decrease in the HOMO-LUMO gaps resulting from the bonding
and antibonding interactions between the monomer units.
The electronic dipole moments of the nonsymmetric tautomer were calculated, and the possibility of charge-separated excited states is discussed.
The relative energies of these tautomers are examined using density functional theory (DFT) calculations for several peripheral substituents.
The relative energies of these tautomers significantly depend on the substituents, and therefore,
the abundance ratios of the three tautomers were affected by the substituents.
The absorption spectra were simulated from the SAC-CI results weighted by the Boltzmann factors obtained from the DFT calculations.
The SAC-CI spectra reproduce the experimental findings well.
The thermal-averaged SAC-CI spectra could explain the observed substituent effect on the structure of the Q-bands in terms of
the relative stabilities and the abundance ratios of the tautomers.
The SAC-CI and time-dependent density functional theory calculations are also compared.
The CAMB3LYP results agreed with the trends of the SAC-CI results;
however, the CAM-B3LYP calculation overestimated the excitation energies in comparison with the SAC-CI and experimental results.
The electronic excited states and electronic absorption spectra of annulated dinuclear free-base phthalocyanine (C58H30N16)
are studied through quantum chemical calculations using the symmetryadapted cluster-configuration interaction (SAC-CI) method.
Three tautomers are possible with respect to the position of the pyrrole protons; therefore, the SAC-CI calculations for these tautomers were performed.
The structures of the Q-band states are discussed based on the character of their molecular orbitals.
The lower energy shift of the Q-bands because of dimerization is explained by the decrease in the HOMO-LUMO gaps resulting from the bonding
and antibonding interactions between the monomer units.
The electronic dipole moments of the nonsymmetric tautomer were calculated, and the possibility of charge-separated excited states is discussed.
The relative energies of these tautomers are examined using density functional theory (DFT) calculations for several peripheral substituents.
The relative energies of these tautomers significantly depend on the substituents, and therefore,
the abundance ratios of the three tautomers were affected by the substituents.
The absorption spectra were simulated from the SAC-CI results weighted by the Boltzmann factors obtained from the DFT calculations.
The SAC-CI spectra reproduce the experimental findings well.
The thermal-averaged SAC-CI spectra could explain the observed substituent effect on the structure of the Q-bands in terms of
the relative stabilities and the abundance ratios of the tautomers.
The SAC-CI and time-dependent density functional theory calculations are also compared.
The CAMB3LYP results agreed with the trends of the SAC-CI results;
however, the CAM-B3LYP calculation overestimated the excitation energies in comparison with the SAC-CI and experimental results.
|
| 23. |
Aerobic oxidation of methanol to formic acid on Au20-:
a theoretical study on the reaction mechanism
K. Bobuatong, S. Karanjit, R. Fukuda, M. Ehara, and H. Sakurai, Phys. Chem. Chem. Phys. 14, 3103-3111 (2012) + [Abstract] 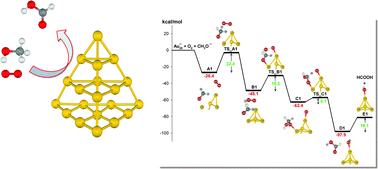 The aerobic oxidation of methanol to formic acid catalyzed by Au20 has been investigated quantum chemically using density functional theory with the M06 functional.
Possible reaction pathways are examined taking account of full structure relaxation of the Au20 cluster.
The proposed reaction mechanism consists of three elementary steps: (1) formation of formaldehyde from methoxy species activated by a superoxo-like anion on the gold cluster;
(2) nucleophilic addition by the hydroxyl group of a hydroperoxyl-like complex to formaldehyde resulting in a hemiacetal intermediate;
and (3) formation of formic acid by hydrogen transfer from the hemiacetal intermediate to atomic oxygen attached to the gold cluster.
A comparison of the computed energetics of various elementary steps indicates that C-H bond dissociation of the methoxy species
leading to formation of formaldehyde is the rate-determining step.
A possible reaction pathway involving single-step hydrogen abstraction, a concerted mechanism, is also discussed.
The stabilities of reactants, intermediates and transition state structures are governed by the coordination number of the gold atoms,
charge distribution, cooperative effect and structural distortion, which are the key parameters for understanding the relationship between
the structure of the gold cluster and catalytic activity in the aerobic oxidation of alcohols.
The aerobic oxidation of methanol to formic acid catalyzed by Au20 has been investigated quantum chemically using density functional theory with the M06 functional.
Possible reaction pathways are examined taking account of full structure relaxation of the Au20 cluster.
The proposed reaction mechanism consists of three elementary steps: (1) formation of formaldehyde from methoxy species activated by a superoxo-like anion on the gold cluster;
(2) nucleophilic addition by the hydroxyl group of a hydroperoxyl-like complex to formaldehyde resulting in a hemiacetal intermediate;
and (3) formation of formic acid by hydrogen transfer from the hemiacetal intermediate to atomic oxygen attached to the gold cluster.
A comparison of the computed energetics of various elementary steps indicates that C-H bond dissociation of the methoxy species
leading to formation of formaldehyde is the rate-determining step.
A possible reaction pathway involving single-step hydrogen abstraction, a concerted mechanism, is also discussed.
The stabilities of reactants, intermediates and transition state structures are governed by the coordination number of the gold atoms,
charge distribution, cooperative effect and structural distortion, which are the key parameters for understanding the relationship between
the structure of the gold cluster and catalytic activity in the aerobic oxidation of alcohols.
|
| 22. |
Photophysical properties and photochemistry of EE-, ZE-, and ZZ-
1,4-dimethoxy-2,5-bis [2-(thien-2-yl)ethyenyl] benzene in solution:
theory and experiment
S. Suramitr, S. Phalinyot, P. Wolschann, R. Fukuda, M. Ehara, and S. Hannongbua, J. Phys. Chem. A 116, 924-937 (2012) + [Abstract] 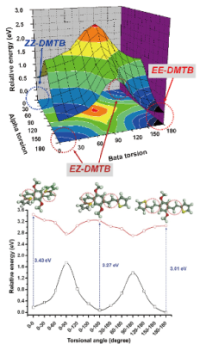 Photophysical properties and photoisomerization of 1,4-dimethoxy-2,5-bis[2-(thien-2-yl)ethenyl] benzene (DMTB) have been investigated
for the EE-, EZ-, and ZZ- stereoisomers.
The EE-DMTB was prepared, and the absorption/fluorescence spectra of EE-isomer as well as transient spectra in photoisomerization among three isomers were observed.
Absorption and fluorescence spectra of three isomers were analyzed by the symmetry-adapted cluster-configuration interaction (SAC-CI)
and time-dependent density functional theory (TDDFT) methods.
The characteristics of the absorption spectra of three isomers were satisfactorily reproduced by the direct SAC-CI and TDDFT methods in both peak position and intensity.
The relative stability of three isomers and the photoisomerization among these isomers were also examined theoretically.
The ground (S0) and first excited state (S1) geometries were calculated by the DFT/TDDFT method with the M06HF functional,
and the calculated S0 structures of EE- and ZZ- isomers agreed well with those of the X-ray structures.
The geometry relaxation in the S1 state was interpreted with regard to the excitation character.
The solvent effect in the absorption and fluorescence spectra was examined by the polarizable continuum model (PCM) and was found to be 0.05 0.20 eV, reflecting the charge polarization.
The results show that the photophysical properties of DMTB can be controlled with the conformation constraint and also indicate the possibility
of a photofunctional molecular device such as a switching function.
Photophysical properties and photoisomerization of 1,4-dimethoxy-2,5-bis[2-(thien-2-yl)ethenyl] benzene (DMTB) have been investigated
for the EE-, EZ-, and ZZ- stereoisomers.
The EE-DMTB was prepared, and the absorption/fluorescence spectra of EE-isomer as well as transient spectra in photoisomerization among three isomers were observed.
Absorption and fluorescence spectra of three isomers were analyzed by the symmetry-adapted cluster-configuration interaction (SAC-CI)
and time-dependent density functional theory (TDDFT) methods.
The characteristics of the absorption spectra of three isomers were satisfactorily reproduced by the direct SAC-CI and TDDFT methods in both peak position and intensity.
The relative stability of three isomers and the photoisomerization among these isomers were also examined theoretically.
The ground (S0) and first excited state (S1) geometries were calculated by the DFT/TDDFT method with the M06HF functional,
and the calculated S0 structures of EE- and ZZ- isomers agreed well with those of the X-ray structures.
The geometry relaxation in the S1 state was interpreted with regard to the excitation character.
The solvent effect in the absorption and fluorescence spectra was examined by the polarizable continuum model (PCM) and was found to be 0.05 0.20 eV, reflecting the charge polarization.
The results show that the photophysical properties of DMTB can be controlled with the conformation constraint and also indicate the possibility
of a photofunctional molecular device such as a switching function.
|
| 21. |
Excited-state geometry and vibrational frequency studied by the analytical energy gradients of the SAC-CI method I,
application to HAX type molecules
M. Ehara, F. Oyagi, Y. Abe, R. Fukuda, and H. Nakatsuji, J. Chem. Phys. 135, 044316-1--14 (2011) + [Abstract]
In this series of studies, we systematically apply the analytical energy gradients of the direct symmetry-adapted cluster-configuration interaction
singles and doubles nonvariational method to calculate the equilibrium geometries and vibrational frequencies of excited and ionized states of molecules.
The harmonic vibrational frequencies were calculated using the second derivatives numerically computed from the analytical first derivatives
and the anharmonicity was evaluated from the three-dimensional potential energy surfaces around the local minima.
In this paper, the method is applied to the low-lying valence singlet and triplet excited states of HAX-type molecules, HCF, HCCl, HSiF, HSiCl, HNO, HPO, and their deuterium isotopomers.
The vibrational level emission spectra of HSiF and DSiF and absorption spectra of HSiCl and DSiCl were also simulated within the Franck-Condon approximation
and agree well with the experimental spectra.
The results show that the present method is useful and reliable for calculating these quantities and spectra.
The change in geometry in the excited states was qualitatively interpreted in the light of the electrostatic force theory.
The effect of perturbation selection with the localized molecular orbitals on the geometrical parameters and harmonic vibrational frequencies is also discussed.
|
| 20. |
Symmetry and vibrationally resolved absorption spectra near the N K edges of N2O: Experiment and theory
M. Ehara, T. Horikawa, R. Fukuda, H. Nakatsuji, T. Tanaka, H. Kato, M. Hoshino, H. Tanaka, R. Feifel, and K. Ueda, Phys. Rev. A 83, 062506-1--12 (2011) + [Abstract]
In this study, angle-resolved energetic-ion yield spectra were measured in the N 1s excitation region of N2O.
A Franck-Condon analysis based on ab initio two-dimensional potential energy surfaces of the core-excited Rydberg states,
which were calculated by the symmetry-adapted cluster-configuration interaction method, reproduced observed vibrational excitations
specific to the individual Rydberg states well and enabled quantitative assignments.
Geometric changes in the terminal nitrogen Nt 1s and the central nitrogen Nc 1s excited states
with respect to the 3pπ, 3pσ, and 4sσ transitions were analyzed.
The coupling of these valence and Rydbergs states was examined based on the second moment analysis.
Irregular Rydberg-state behavior in the Nc 1s-1 4sσ state was observed.
|
| 19. |
Nonequilibrium solvation for vertical photoemission and photoabsorption processes
using the symmetry-adapted cluster-configuration interaction method in the polarizable continuum model
R. Fukuda, M. Ehara, H. Nakatsuji, and R. Cammi, J. Chem. Phys. 134, 104109-1--11 (2011) + [Abstract]
In this paper, we present the theory and implementation of a nonequilibrium solvation model for the symmetry-adapted cluster (SAC) and symmetry-adapted cluster-configuration interaction (SAC-CI) method in the polarizable continuum model.
For nonequilibrium solvation, we adopted the Pekar partition scheme in which solvent charges are divided into dynamical and inertial components.
With this nonequilibrium solvation scheme, a vertical transition from an initial state to a final state may be described as follows:
the initial state is described by equilibrium solvation, while in the final state, the inertial component remains in the solvation for the initial state; the dynamical component will be calculated self-consistently for the final state.
The present method was applied to the vertical photoemission and absorption of s-trans acrolein and methylenecyclopropene.
The effect of nonequilibrium solvation was significant for a polar solvent.
|
| 18. |
Excited states and electronic spectra of extended tetraazaporphyrins R. Fukuda, M. Ehara, and H. Nakatsuji, J. Chem. Phys. 133, 144316-1--16 (2010) + [Abstract]
Electronic excited states, electronic absorption, and magnetic circular dichroism (MCD) spectra of free-base tetraazaporphyrin (TAP), phthalocyanine (Pc), naphthalocyanine (Nc), and anthracocyanine (Ac)
were studied by quantum chemical calculations using the symmetry-adapted cluster-configuration interaction (SAC-CI) method.
Not only optically allowed states including the Q- and B-bands but also optically forbidden states were calculated for transitions whose excitation energies were lower than 4.5 eV.
The present SAC-CI calculations consistently assigned the absorption and MCD peaks as optically allowed π → π* excitations, although these calculations using double-zeta basis limit quantitative agreement and discussion.
For Nc and Ac, excited states beyond the four-orbital model appeared in the low-energy region.
The low-energy shifts of the Q-bands with the extension of molecular size were explained by the orbital energies. The splitting of the Q-bands decreases with extension of the molecular size.
This feature was reproduced by the SAC-CI calculations but the configuration interaction with single excitations and time-dependent density functional theory calculations failed to reproduce this trend.
Electron correlation in the excited states is important in reproducing this splitting of the Q-bands and in describing the energy difference between the B2u and B3u states of free-base porphyrins.
|
| 17. |
Symmetry-adapted cluster and symmetry-adapted cluster-configuration interaction method in the polarizable continuum model:
Theory of the solvent effect on the electronic excitation of molecules in solution R. Cammi, R. Fukuda, M. Ehara, and H. Nakatsuji, J. Chem. Phys. 133, 024104-1--24 (2010) + [Abstract]
In this paper we present the theory and implementation of the symmetry-adapted cluster (SAC) and symmetry-adapted cluster-configuration interaction (SAC-CI) method,
including the solvent effect, using the polarizable continuum model (PCM). The PCM and SAC/SAC-CI were consistently combined in terms of the energy functional formalism.
The excitation energies were calculated by means of the state-specific approach, the advantage of which over the linear-response approach has been shown.
The single-point energy calculation and its analytical energy derivatives are presented and implemented, where the free-energy and its derivatives are evaluated because of the presence of solute-solvent interactions.
We have applied this method to s-trans-acrolein and metylenecyclopropene of their electronic excitation in solution.
The molecular geometries in the ground and excited states were optimized in vacuum and in solution, and both the vertical and adiabatic excitations were studied.
The PCM-SAC/SAC-CI reproduced the known trend of the solvent effect on the vertical excitation energies but the shift values were underestimated.
The excited state geometry in planar and nonplanar conformations was investigated. The importance of using state-specific methods was shown for the solvent effect on the optimized geometry in the excited state.
The mechanism of the solvent effect is discussed in terms of the Mulliken charges and electronic dipole moment.
|
| 16. |
Valence ionized states of iron pentacarbonyl and η5-cyclopentadienyl cobalt dicarbonyl studied by symmetry-adapted cluster-configuration interaction calculation and collision-energy resolved Penning ionization electron spectroscopy R. Fukuda, M. Ehara, H. Nakatsuji, N. Kishimoto, and K. Ohno, J. Chem. Phys. 132, 084302-1--12 (2010) + [Abstract]
Valence ionized states of iron pentacarbonyl Fe(CO)5 and η5-cyclopentadienyl cobalt dicarbonyl Co(η5-C5H5)(CO)2
have been studied by ultraviolet photoelectron spectroscopy, two-dimensional Penning ionization electron spectroscopy (2D-PIES), and symmetry-adapted cluster-configuration interaction calculations.
Theory provided reliable assignments for the complex ionization spectra of these molecules, which have metal-carbonyl bonds.
Theoretical ionization energies agreed well with experimental observations and the calculated wave functions could explain the relative intensities of PIES spectra.
The collision-energy dependence of partial ionization cross sections (CEDPICS) was obtained by 2D-PIES.
To interpret these CEDPICS, the interaction potentials between the molecules and a Li atom were examined in several coordinates by calculations.
The relation between the slope of the CEDPICS and the electronic structure of the ionized states, such as molecular symmetry and the spatial distribution of ionizing orbitals, was analyzed.
In Fe(CO)5, an attractive interaction was obtained for the equatorial CO, while the interaction for the axial CO direction was repulsive.
For Co(η5-C5H5)(CO)2, the interaction potential in the direction of both Co—C—O and Co—Cp ring was attractive.
These anisotropic interactions and ionizing orbital distributions consistently explain the relative slopes of the CEDPICS.
|
| 15. |
Valence ionization spectra of group six metal hexacarbonyls studied
by the symmetry-adapted cluster-configuration interaction method R. Fukuda, S. Hayaki, and H. Nakatsuji, J. Chem. Phys. 131, 174303-1--10 (2009) + [Abstract]
The valence ionization spectra up to 20 eV of group six metal carbonyls, chromium hexacarbonyl, molybdenum hexacarbonyl, and tungsten hexacarbonyl were studied by the symmetry-adapted cluster-configuration interaction (SAC-CI) method.
The assignments of the spectra are given based on reliable SAC-CI calculations.
The relativistic effects including the spin-orbit effects are important for the ionization spectrum of W(CO)6.
The relation between the metal-CO distance and ionization energies was examined.
The lowest ionization energies of the three metal carbonyls are approximately the same because of the energy dependence of the metal-CO length and relativistic effects.
In Cr(CO)6, the Cr—CO interaction significantly increases the lowest ionization energy in comparison with Mo(CO)6 and W(CO)6 because of the relatively short metal-CO bond length.
The relativistic effect reduces the lowest ionization energy of W(CO)6 because the effective core potential of 5d electrons is more efficiently screened as a result of the relativistic contraction of the inner electrons.
|
| 14. |
Formulation and implementation of direct algorithm for the symmetry adapted cluster
and symmetry adapted cluster-configuration interaction method R. Fukuda and H. Nakatsuji, J. Chem. Phys. 128, 094105-1--14 (2008) + [Abstract]
We present a new computational algorithm, called direct algorithm, for the symmetry-adapted cluster (SAC) and SAC-configuration interaction (SAC-CI) methodology for the ground, excited, ionized, and electron-attached states.
The perturbation-selection technique and the molecular orbital index based direct sigma-vector algorithm were combined efficiently with the use of the sparse nature of the matrices involved.
The formal computational cost was reduced to O(N2 × M) for a system with N-active orbitals and M-selected excitation operators.
The new direct SAC-CI program has been applied to several small molecules and free-base porphin and has been shown to be more efficient than the conventional nondirect SAC-CI program for almost all cases.
Particularly, the acceleration was significant for large dimensional computations. The direct SAC-CI algorithm has achieved an improvement in both accuracy and efficiency.
It would open a new possibility in the SAC/SAC-CI methodology for studying various kinds of ground, excited, and ionized states of molecules.
|
| 13. |
Vibration-induced suppression of valence-Rydberg mixing in the O1s -> ns-σ Rydberg series in N2O T. Tanaka, M. Hoshino, H. Kato, M. Ehara, N. Yamada, R. Fukuda, H. Nakatsuji, Y. Tamenori, J. R. Harries, G. Pruemper, H. Tanaka, and K. Ueda, Phys. Rev. A 77, 012709-1--4 (2008) + [Abstract]
Symmetry-resolved ion-yield spectroscopy of vibrationally excited N2O in the vicinity of the O 1s ionization
threshold has been carried out using an angle-resolved ion detection technique. The spectral intensity of
the O 1s → nsσ Rydberg series, which has σ* valence character, is significantly reduced by the excitation of the
bending vibration in the electronic ground state. Using an ab initio analysis of the electronic part of the second
moment <r2>, this suppression is interpreted as being due to a decrease in the mixing of the valence character
in the nsσ Rydberg states with decreasing bond angle.
|
| 12. |
Spectroscopy of sodium atom in liquid helium cluster:
a symmetry adapted cluster -configuration interaction (SAC-CI) study B. Saha, R. Fukuda, H. Nakatsuji and P. K. Mukherjee, Theor. Chem. Acc. 118, 437-441 (2007) |
| 11. |
Symmetry-adapted-cluster/symmetry-adapted-cluster configuration interaction
methodology extended to giant molecular systems: Ring molecular crystals H. Nakatsuji, T. Miyahara, R. Fukuda, J. Chem. Phys. 126, 084104-1--18 (2007) |
| 10. |
Symmetry and vibrationally resolved absorption spectra near the OK edge of N2O: Experiment and theory T. Tanaka, K. Ueda, R. Feifel, L. Karlsson, H. Tanaka, M. Hoshino, M. Kitajima, M. Ehara, R. Fukuda, R. Tamaki, H. Nakatsuji, Chem. Phys. Lett. 435, 182-187 (2007) |
| 9. |
Electronic spectra and photodissociation of vinyl chloride:
A symmetry-adapted cluster configuration interaction study. S. Arulmozhiraja, R. Fukuda M. Ehara, and H. Nakatsuji, J. Chem. Phys. 124, 034312-1--6 (2006) |
| 8. |
Quasirelativistic theory for the magnetic shielding constant.
III. Quasirelativistic second-order Moller-Plesset perturbation theory and its application to tellurium compounds R. Fukuda and H. Nakatsuji, J. Chem. Phys. 123, 044101-1--10 (2005) |
| 7. |
Relativistic configuration interaction and coupled cluster methods using four-component spinors:
Magnetic shielding constants of HX and CH3X (X = F, Cl, Br, I) M. Kato, M. Hada, R. Fukuda, and H. Nakatsuji, Chem. Phys. Lett. 408, 150-156 (2005) |
| 6. |
Quasi-relativistic theory for the magnetic shielding constants.
II. Gauge-including atomic orbitals and applications to molecules R. Fukuda, M. Hada, and H. Nakatsuji, J. Chem. Phys. 118, 1027-1035 (2003) |
| 5. |
Quasi-relativistic theory for the magnetic shielding constants.
I. Formulation of Douglas-Kroll- Hess transformation for the magnetic field and its application to atomic systems R. Fukuda, M. Hada, and H. Nakatsuji, J. Chem. Phys. 118, 1015-1026 (2003) |
| 4. |
Quasirelativistic study of 125Te nuclear magnetic shielding constants and chemical shifts M. Hada, J. Wan, R. Fukuda, and H. Nakatsuji, J. Comput. Chem. 22, 1502-1508 (2001) |
| 3. |
Quasi-relativistic study of 199Hg nuclear magnetic shielding constants of
dimethylmercury, disilylmercury and digermylmercury J. Wan, R. Fukuda, M. Hada, and H. Nakatsuji, J. Phys. Chem. A 105, 128-133 (2001) |
| 2. |
Relativistic effects and the halogen dependencies in the 13C chemical shifts of
CH4-nIn, CH4-nBrn,
CCl4-nIn, and CBr4-nIn (n=0-4) S. Fukawa, M. Hada, R. Fukuda, S. Tanaka, and H. Nakatsuji, J. Comput. Chem. 22, 528-536 (2001) |
| 1. |
Dirac-Fock calculations of the magnetic shielding constants of protons and heavy nuclei in XH2
( X = O, S, Se, and Te): a comparison with quasi-relativistic calculations M. Hada, R. Fukuda, and H. Nakatsuji, Chem. Phys. Lett. 321, 452-458 (2000) |
Review article/Book chapter
| 3. |
The SAC-CI method R. Fukuda and M. Ehara Book series of Japan Society of Coordination Chemistry Vol. 10, Quantum and Computational Chemistry for Metal Complexes, edited by K. Yamaguchi, S. Sakaki, and H. Masuda (Sankyo, Tokyo, 2014) pp. 142-160 (in Japanese) (ISBN 978-4-7827-0709-8) |
| 2. |
Computational materials chemistry for photofunctional molecules based on the highly accurate electronic structure theory M. Ehara and R. Fukuda Expected materials for the future, Vol.12 (No.2), (NTS, Tokyo, 2012) pp. 15-22 (in Japanese) |
| 1. |
Generalized-UHF theory for the magnetic properties with quasi-relativistic Hamiltonian R. Fukuda, M. Hada, and H. Nakatsuji Recent advances in computational chemistry Vol.5, RECENT ADVANCES IN RELATIVISTIC MOLECULAR THEORY, edited by K. Hirao and Y. Ishikawa (World Scientific, Singapore, 2004) pp. 191-220 (ISBN 981-238-709-9) |
Conferene proceedings
| 3. |
Electronic excitation of molecules in solution calculated using the symmetry-adapted cluster?configuration interaction method in the polarizable continuum model
R. Fukuda and M. Ehara AIP Conf. Proc. 1702, 090012 (2015) |
| 2. |
Electronic excited states of macrocyclic compounds: direct SAC-CI study
R. Fukuda, M. Ehara, and H. Nakatsuji, Procedia Computer Science: Proceedings of the International Conference on Computational Science, ICCS 2011 4, 1129-1134 (2011) |
| 1. |
Theoretical spectroscopy of O 1s and N 1s excited states of N2O
M. Ehara, T. Horikawa, R. Fukuda, H. Nakatsuji, T. Tanaka, M. Noshino, H. Tanaka, and K. Ueda, Journal of Physics: Conference Series 288, 012024-1--11 (2011) |
Invited talk/Lecture (international)
| 8. |
Pacifichem 2015, Symposium (#277): Interplay between Theory and Experiment in Catalytic Research, (December 2015, Hawaii, USA) Title of Talk: "Electronic structure and catalytic activity of alloy nanoclusters" |
| 7. |
CECAM workshop "Charge Transfer Modeling in Chemistry: new methods and solutions for a long-standing problem", April 7-10, 2015, Chimie ParisTech, Paris, (France) Title of Talk: "Electronic excitation and charge transfer in polarizable media studied by the symmetry-adapted cluster-configuration interaction (SAC-CI) method" |
| 6. |
11th International Conference of Computational Methods in Science and Engineering (ICCMSE 2015), "Computational Chemistry (CC) Symposium", March 20-23, 2015, Athens (Greece) Title of Talk: "Electronic Excitation of Molecules in Solution Calculated Using the Symmetry-Adapted Cluster Configuration Interaction (SAC-CI) Method in the Polarizable Continuum Model (PCM)" |
| 5. |
93th Annual Meeting of the Chemical Society of Japan, Asian International Symposium - Theoretical Chemistry, Chemoinformatics, Computational Chemistry,
March 24, 2013, Shiga (Japan) Title of Talk: "Electronic excited states of large conjugated molecules studied by the direct SAC-CI method" |
| 4. |
The International Conference on Computational Science (ICCS) 2011, June, 1-3, 2011, Tsukuba (Japan) [moved to Singapore (Singapore)];
workshop "Large Scale Computational Molecular Science" Title of Talk: "Electronic excited states of macrocyclic compounds: direct SAC-CI study" |
| 3. |
Fourth Japan-Czech-Slovak Symposium for Theoretical and Computational Chemistry, May 18-20, 2011, Liblice (Czech republic) Title of Talk: "Development and Applications of Direct SAC-CI Method" (Invited talk) |
| 2. |
Fifth International Conference on Porphyrins and Phthalocyanines, July 6-11, 2008, Moscow (Russia) Title of Talk: "Theoretical spectroscopy of porphyrins, porphyrazines, and phthalocyanines by the SAC-CI method" (Invited talk) |
| 1. |
Symposium of "Recent Trends in Many-Body Methods for Electronic Structure and Properties of
Atoms and Molecules" January 11-13, 2007, Bhubaneswar and Puri (India) Title of talk: "Relativistic effect on magnetic shielding constants and SAC-CI calculation of hyperfine splitting constants", (Invited talk) |
Contributed talk (international)
| 2. |
The Seventh Congress of the International Society for Theoretical Chemical Physics (ISTCP-VII), September 2-8, 2011, Waseda, (Tokyo, Japan) Title of Talk: "Electronic excited states of large conjugated molecules" |
| 1. |
Pacifichem 2010, Symposium #138: Molecular Theory for Real Systems and Chemical Reactions, December 15-20, 2010, Honolulu (Hawaii, USA) Title of Talk: "Symmetry-adapted cluster and symmetry-adapted cluster-configuration interaction method in the polarizable continuum model - theory of the solvent effect on the electronic excitation of molecules in solution" |